Патологія крові та органів кровотворення. Хвороби крові та органів кровотворення Захворювання крові та кровотворних органів
А-Я А Б В Г Д Е Ж З І Й К Л М Н О П Р С Т У Ф Х Ц Ч Ш Щ Е Ю Я Усі розділи Спадкові хворобиНевідкладні стани Очні хворобиДитячі хвороби Чоловічі хвороби Венеричні хвороби Жіночі хвороби Шкірні хворобиІнфекційні хвороби Нервові хвороби Ревматичні хворобиУрологічні хвороби Ендокринні хвороби Імунні хвороби Алергічні хворобиОнкологічні хвороби Хвороби вен та лімфовузлів Хвороби волосся Хвороби зубів Хвороби крові Хвороби молочних залоз Хвороби ОДС та травми Хвороби органів дихання Хвороби органів травлення Хвороби серця та судин Хвороби товстого кишечника Хвороби вуха, горла, носа Наркологічні проблеми Психічні розладиПорушення мови Косметичні проблемиЕстетичні проблеми
Хвороби крові становлять численну та гетерогенну групу синдромів, що розвиваються при порушеннях якісного та кількісного складу крові. Практичним напрямом, що розробляє принципи діагностики та лікування хвороб крові, є гематологія та її окрема гілка – онкогематологія. Фахівці, які здійснюють корекцію стану крові та кровотворних органів, називаються гематологами. Найбільш тісними міждисциплінарними зв'язками гематологія пов'язана із внутрішніми хворобами, імунологією, онкологією, трансфузіологією.
З давніх-давен у багатьох культурах людська кров наділялася містичними властивостями, символізувала божественне джерело і потік життя. «Дорогоцінна», «гаряча», «невинна», «молода», «царська» - якими тільки властивостями не наділяли люди кров, а епітет «кровний» завжди означав вищий ступінь тих чи інших проявів - кровна спорідненість, кровний ворог, кровна образа , кровна помста.
Тим часом, з погляду фізіології, кров являє собою рідке середовище організму, що безперервно циркулює за системою судин і виконує ряд найважливіших функцій - дихальну, транспортну, регуляторну, захисну та ін. Кров складається з рідкої фракції - плазми та зважених у ній формених елементів– клітин крові (еритроцитів, тромбоцитів та лейкоцитів). До основних органів кровотворення (гемоцитопоезу), що є своєрідною «фабрикою» з виробництва клітин крові, відносяться червоний кістковий мозок, тимус, лімфоїдна тканина та селезінка. Про хвороби крові говорять у разі порушення морфології чи кількості тих чи інших клітин крові чи зміни властивостей плазми.
Усі хвороби крові та кровотворної системи класифікуються, виходячи з ураження того чи іншого її компонента. У гематології хвороби крові прийнято поділяти на три великі групи: анемії, гемобластози та гемостазіопатії. Так, до частих аномалій та уражень еритроцитів відносяться дефіцитні, гемолітичні, гіпо-і апластичні анемії. У структуру гемобластозів входять лейкози та гематосаркоми. Хвороби крові, пов'язані з ураженням системи гемостазу (гемостазіопатії) включають гемофілію, хворобу Віллебранда, тромбоцитопатії, тромбоцитопенії, ДВС-синдром та ін.
Хвороби крові можуть мати вроджений та набутий характер. Вроджені захворювання (серповидно-клітинна анемія, таласемія, гемофілія та ін) пов'язані з генними мутаціями або хромосомними аномаліями. Розвиток набутих хвороб крові може бути спровоковано численними середовищними факторами: гострою та хронічною крововтратою, впливом на організм іонізуючої радіації або хімічних агентів, вірусними інфекціями (краснухою, кір, епідемічним паротитом, грипом, інфекційним мононуклеозом). арною недостатністю, порушенням всмоктування поживних речовин та вітамінів у кишечнику та ін. При проникненні в кров бактеріальних або грибкових агентів виникає тяжке захворювання крові інфекційного генезу – сепсис. Багато хвороб крові йдуть рука об руку з колагенозами.
Прояви хвороб крові багатоликі і не завжди специфічні. Характерними ознаками анемії є безпричинна стомлюваність і слабкість, запаморочення до непритомності, задишка при фізичних навантаженнях, блідість шкірних покривів. Порушення згортаючої ланки крові характеризуються петехіальними крововиливами та екхімозами, різного родукровотечами (ясенними, носовими, матковими, шлунково-кишковими, легеневими та ін). У клініці лейкозів першому плані виступають інтоксикаційний чи геморагічний синдроми.
Найпершим методом, з якого починається діагностика хвороб крові, є дослідження гемограми ( клінічного аналізу) з визначенням кількісного складу крові та морфології формених елементів. При хворобах крові, що протікають з порушенням гемостазу, досліджується кількість тромбоцитів, час зсідання крові та кровотечі, протромбіновий індекс, коагулограма; проводяться різноманітних проби - проба джгута, щипка, банкова проба та інших.
Для оцінки змін, що відбуваються у кровотворних органах, використовується пункція кісткового мозку(стернальна пункція. У рамках з'ясування причин анемічного синдрому пацієнтам може знадобитися консультація гастроентеролога та гінеколога; проведення ФГДС, колоноскопії, УЗД печінки).
Будь-які зміни гемограми або мієлограми, а також симптоми, що вказують на ймовірність розвитку хвороб крові, вимагають компетентної оцінки з боку гематолога, проведення динамічного спостереженнячи спеціалізованого лікування під його контролем. Сучасна гематологія виробила основні принципи терапії різних хвороб крові та накопичила величезний досвід у їх лікуванні. По можливості лікування хвороб крові починають з усунення причин і факторів ризику, корекції роботи внутрішніх органів, заповнення відсутніх речовин і мікроелементів (заліза - при залізодефіцитній анемії, вітаміну В12 - при В12-дефіцитної анемії, фолієвої кислоти- При фолієводефіцитної анемії і т. д.).
У деяких випадках може бути показаний прийом кортикостероїдів, гемостатичних препаратів, проведення екстракорпоральної гемокорекції (, еритроцитаферезу). Нерідко гематологічні хворі потребують переливання крові та її компонентів. Найактуальнішими та ефективними на сьогоднішній день методами лікування гематобластозів у всьому світі є цитостатична терапія, радіотерапія, алогенна та аутологічна трансплантація кісткового мозку, введення стовбурових клітин. Цілий рядхвороб крові (тромбоцитопенічна пурпура, аутоімунні анемії, мієлолейкоз та ін.) є показанням для видалення селезінки – спленектомії. Лікування хвороб крові в Москві проводиться у спеціалізованих гематологічних медико-наукових центрах, що мають технічне оснащення світового рівня та висококваліфікованих фахівців.
» Ви зможете знайти відповіді на основні питання щодо хвороб крові, познайомитися з основними нозологічними формами, симптоматикою, принципами діагностики та лікування.
Захворювання крові та кровотворних органів є частою причиноюранньої смерті дітей та дорослих. Це тим, що патологічні процеси у плазмі важко піддаються лікуванню, яке ще й дуже дороге. Своєчасна терапія та профілактичні заходидопомагають знизити ризик можливої інвалідності або смерті.
Хвороби крові дуже небезпечні для людини
Класифікація захворювань крові
Патогенезом хвороб крові та кровотворних органів займається наука гематологія (haematologia).
Залежно від етіології виникнення та класу, виділяють основні види порушень:
- хвороби еритроцитів;
- патології лейкоцитів;
- захворювання тромбоцитів;
- Незгортання крові - геморагічні діатези.
Хвороби, спричинені зміною у червоних кров'яних тільцях
Зміна кількості або якості еритроцитів у плазмі завжди свідчить про погіршення основних функцій крові. Надлишок таких білків () зустрічається рідко, набагато частіше люди стикаються з їхнім дефіцитом ().
Основними причинами анемії є:
- кровотечі різної тяжкості - великі втрати крові в результаті травми, операції або незначні, але часті, які пов'язані з носовими кровотечами, рясними місячними, виразками, що кровоточать в травній системі;
- швидкий розпад еритроцитів через їх слабку або деформовану оболонку;
- збої в метаболізмі червоних кров'яних клітин та гемоглобіну, що провокує підвищений виробітокехіноцитів (старі еритроцити).
Відхилення у червоних кров'яних клітинах спричиняє порушення основних функцій таких тілець.
На цьому фоні розвиваються небезпечні хвороби:
- Гостра чи хронічна геморагічна анемія.
- Генетична анемія гемолітичного характеру – серповидно-клітинна анемія, таласемія, сфероцитоз, еліптоцитоз, акантоцитоз.
- Набута недокрів'я внаслідок аутоімунних відхилень крові, патологій дрібних та середніх судин, гемолітико-уремічного синдрому, малярії, гемолітичної інтоксикації.
- Дефіцитна анемія.
- Порфирії (ураження клітинних елементів, які беруть участь в утворенні гемоглобіну).
- Апластична анемія (відхилення у роботі кісткового мозку).

Кров'яні тільця здорової кровіта при анемії
Зменшення кількості крові або недокрів'я може бути спричинене посиленою витратою гемоглобіну. Таке спостерігається під час вагітності, годування груддю, а також у спортсменів.
Незалежно від патогенезу, основними симптомами анемії бувають:
- швидка стомлюваність, постійна слабкістьта часті запаморочення;
- блідий колір шкірних покривів;
- прискорене серцебиття, шум у вухах;
- погіршення пам'яті, працездатності, порушення сну;
- колір губ і ясен стає світло-рожевим, біліють вушні раковини (у дітей).

При анемії з'являється шум у вухах
Рідкісні захворювання крові можуть виникати при підвищеному продукуванні еритроцитів. Зазвичай червоний відросток не уражається злоякісними пухлинами, і всі патології, що виникли внаслідок збільшення кількості кров'яних тілець, мають доброякісний перебіг.
- Поліцитемія (плеторичний синдром) – у плазмі підвищуються не лише еритроцити, а й інші клітини (тромбоцити, лейкоцити). У цьому збільшується загальний обсяг крові.
- Еритремія – підвищене продукування лише червоних кров'яних тілець.
- Хвороба Вакеза – зачіпає всі паростки кровотворення, особливо еритроїдні, є доброякісним лейкозом.

Поліцитемія - збільшення кількості кров'яних тілець
Яскравими ознаками хвороб збільшеного рівня еритроцитів виступають такі стани:
- почервоніння шкіри на обличчі;
- часті припливи жару;
- свербіж і печіння по всьому тілу, яке стає більш помітним після теплої ванничи душа;
- больові напади у стопах;
- відчуття печіння у фалангах верхніх кінцівок.

При збільшенні еритроцитів з'являється почервоніння на шкірі.
Порушення якості та кількості еритроцитів у плазмі вже на ранніх стадіях проявляється неприємними симптомами. Тому чим швидше діагностовано захворювання, тим легше з ним боротися.
Хвороби крові, пов'язані з відхиленнями у лейкоцитах
Білі кров'яні клітини виробляються у кістковому мозку. Це відповідь імунітету як проліферативного запалення проникнення в організм інфекції чи вірусів. Лейкоцити представлені 5 основними формами – еозинофіли, моноцити, нейтрофіли, базофіли. Порушення лейкоцитарної гілки кровотворення має пухлинний характер і нерідко викликає рак.
Спровокувати зміни в білих тільцях можуть такі причини, як:
- вплив інфекцій та вірусів;
- отруєння хімічними речовинами;
- дефекти в паростку, які закладені на генетичному рівні;
- радіаційний вплив;
- вплив кортикостероїдів (провокують підвищення білих клітин).
Внаслідок впливу зовнішніх та внутрішніх факторів лейкоцити або перестають нормально вироблятися, через що спостерігається їх дефіцит (), або аномально зростати (лейкоцитоз). Інтенсивне збільшення білих кров'яних тілець може бути спровоковано вивільненням незрілих або дефектних клітин кісткового мозку.
Залежно від порушення функцій певного типу клітин лейкоцитів чи їхньої сукупності розвиваються конкретні захворювання.
- Лімфоцитопенія – критичне зменшення лімфоцитів.
- Лімфоцитарний лейкоз, лімфома або лімфогранулематоз – інтенсивний поділ лімфоцитів. Захворювання супроводжується сильним збільшенням лімфатичних вузліводночасно у кількох частинах тіла.
- Нейтропенія - слабка вироблення нейтрофілів.
- Нейтрофільний лейкоцитоз – неконтрольоване підвищення нейтрофілів у плазмі.
- Інфекційний мононуклеоз – інфекційне захворювання крові, спричинене вірусом герпесу.

При лімфоцитопенії зменшується кількість лейкоцитів
Всі лейкози та лімфоми носять злоякісний характер та являють собою онкогемологічні захворювання, які здатні проявлятися як у дітей, так і у дорослих. Виходячи із стадії, на який почали розвиватися порушення в лейкоцитарних паростках, патології мають гостре чи хронічне перебіг.
Основними симптомами ураження лейкоцитів виступають:
- часті головні та суглобові болі, прогресуючий дискомфорт у кістках (оссалгічний синдром);
- кровоточивість ясен, больові відчуття у роті, неприємний запах;
- почуття слабкості, втоми, частий озноб та незначне підвищення температури тіла;
- погіршення пам'яті, зниження працездатності;
- біль у ротовій порожнині та горлі, що посилюється під час їжі та прийому рідини;
- помітне збільшення лімфовузлів.

Збільшення лімфовузлів відбувається при зменшенні лімфоцитів
При розвитку лімфом кістковий мозок уражається на останніх стадіях, коли починаються процес метостаз. А ось лейкози провокують патологічні зміни ще на початку захворювання, тому що продукують у крові величезну кількість бластів (нехарактерних клітин).
Захворювання тромбоцитів
Без'ядерні кров'яні клітини відповідають за нормальну консистенцію крові та перешкоджають неконтрольованим кровотечам (утворюють тромби).
Відхиленнями у нормальному функціонуванні тромбоцитів є такі стани:
- дефекти (спадкові або набуті) у структурі клітин, що заважають їхній нормальній роботі (тромбоцитопатія);
- критичне зменшення без'ядерних клітин (тромбоцитопенія);
- підвищений виробіток тромбоцитів (тромбоцитоз).
Найчастіше зустрічається тромбоцитопенія, для якої характерно зменшення вироблення тромбоцитів або їх швидке руйнування.
Подібний патогенез властивий наступним захворюванням:
- Аллоімунна пурпура новонароджених.
- Судинна псевдогемофілія.
- Хвороба Верльгофа (ідіопатична тромбоцитопенічна пурпура).
- Трансімунні пурпури новонароджених.
- Синдром Евансу.

Алергічний васкуліт характерний зменшенням кількості тромбоцитів
Нерідко низька вироблення тромбоцитів супроводжується дефектами їх структур та зниженням виконуваних функцій. У такому випадку можливий розвиток патологічних синдромів (TAR, Бернара-Сульє, Мая-Хегглина, Чідака-Хігасі) та хвороб (Віскота-Олдріча, Віллебранда, Херманського-Пудлака, тромбластенія Гланцмана).
Явними ознаками хвороб тромбоцитів є:
- крововиливи під шкіру – синці чи екхімози;
- кровотеча з ясен при чищенні зубів;
- червоні плями на шкірних покривах (пурпура чи підшкірні крововиливи);
- дрібні кров'яні плями плоскої форми на нижніх кінцівках (петехії);
- часті кровотечі з носа, рясні місячні.

При хворобах тромбоцитів з'являються крововиливи під шкірою
Порушення функцій тромбоцитів може спричинити спонтанні, рясні внутрішні чи зовнішні кровотечі. Тому важливо не ігнорувати неприємні симптоми, а при появі відразу звертатися до лікарні.
Геморагічні діатези
Негативні відхилення в згортання крові провокують розвиток цілого списку захворювань, об'єднаних в одну групу - геморагічні діатези. Головною особливістю таких патологій є підвищена схильність людини до рясних крововтрат.
Найчастішими провокуючими факторами незгортання є:
- генетичні дефекти у структурних елементах кровотворення, що передаються у спадок (вроджені відхилення);
- порушення в цілісності стінок кровоносних судин, яке відбулося в результаті розвитку супутніх хвороб (придбаний геморагічний діатез);
- зміни у тромбоцитарному гемостазі (збільшення чи зменшення тромбоцитів, дефекти клітинних оболонок).
Хвороби крові, що входять до групи геморагічних діатезів:
- Гемофілія (страждають лише представники чоловічої статі). Характерні риси – спонтанні крововиливиу м'які тканини (суглоби, м'язи), розвиток великих гематом на тілі.
- Гемангіоми (доброякісні пухлини).
- Геморагічний васкуліт.
- Синдроми (Казабаха-Меррітта, Гассера, Луї-Бар).
- Тромботична тромбоцитопенічна пурпура.
- Придбані коагулопатії – афібриногенемія, фібринолітична кровоточивість.
Проблеми зі зсідання крові проявляються різним висипанням на тілі, яке у важких формах може перетворюватися на виразкові ураження.
Супутніми ознаками геморагічного діатезу є:
- нудота, болючі відчуття в ділянці живота;
- блювання з кров'яними домішками;
- синці та гематоми по всьому тілу;
- регулярні кровотечі з носа, рота, органів травного тракту;
- запаморочення, мігрені, слабкість;
- блідість шкірних покривів.

Анемія характерна частими кровотечамиз носа
Пацієнт із патологіями крові може страждати від кількох проявів захворювання або відчувати їх у сукупності. Все залежить від стадії та виду конкретної хвороби.До якого лікаря звернутись?
Якщо у людини з'являються гематоми і синці по тілу, тривалий час тримається висока температура, збільшені лімфовузли, дуже бліда шкіра або спостерігається схильність до крововтрат, потрібно негайно звернутися до лікаря. Первинною діагностикою патологічних процесів крові, і навіть їх лікуванням займається . Фахівець допомагає знайти причину захворювання, підібрати адекватну терапію та подальшу профілактику.
Діагностика
Для визначення захворювання крові, яке спричинило погіршення стану пацієнта, фахівець може призначити інструментальні та лабораторні методи дослідження.
Для цього людині потрібно здати біологічний матеріал та пройти апаратний моніторинг:
- - Вивчається стан всіх кровотворних клітин.
- - Дослідження маркерів згортання крові.
- Гістологія та біопсія лімфовузлів – виявлення патогенезу негативних процесів.
- Морфологічне дослідження кісткового мозку в комплексі зі стернальною пункцією – вивчення активності паростків кровотворення та виявлення у тканинах злоякісних пухлин.
- Комп'ютерна томографія – моніторинг внутрішніх органів прокуратури та виявлення у яких руйнівних процесів.
- УЗД – використовується для огляду лімфатичних вузлів та органів черевної порожнини.

Для визначення стану кров'яних тілець проводиться аналіз крові
Комплексне обстеження дозволяє з високою точністю поставити діагноз і зробити прогноз захворювання. Це допоможе зорієнтувати людину на майбутнє лікування та підібрати найбільш ефективну терапію.
Лікування
Боротьба із захворюваннями крові – тривалий процес, який вимагає комплексного підходу. У ході лікування можуть використовуватися консервативні методи або хірургічне втручання(залежно від виду патології та ступеня занедбаності).
Медикаменти
Основна мета терапії препаратами – покращити стан хворого та відновити нормальне функціонування крові. Залежно від виду хвороби та її тяжкості лікар підбирає індивідуальне лікування конкретної патології.
Універсального методу для всіх хвороб крові немає, але є найчастіше вживані групи ліків, які використовуються в тому чи іншому випадку:
- Антикоагулянти - Аспірин Кардіо, Плогрель, Стрептаза, Аспігрель.
- Гемостатичні препарати - Амінокапронова кислота, Імунат, Апротекс, Вікасол, Транексам.
- Антианемічні засоби - Аскофол, Гемофер, Фолієва кислота, Заліза глюконат 300, Хеферол.
- Плазмозамінні розчини - Альбумін сироватковий, Амінокровін, Кабівен периферичний, Поліглюкін.

Імунат – гемостатичний препарат
Дозування та тривалість терапії визначає лікар. Зазвичай лікування відбувається у амбулаторних умовах, винятком виступають важкі пухлинні патології крові, коли госпіталізація життєво необхідна.
Немедикаментозне лікування
Небезпечні патології кровотворних систем (лейкози, лімфоми) не піддаються консервативній терапії.
Для боротьби зі злоякісними пухлинами застосовують такі ефективні методи, як:
- пересадка кісткового мозку (стовбурових клітин);
- хіміотерапія;
- переливання компонентів крові.

Для боротьби зі злоякісними пухлинами застосовується переливання крові
Можливі ускладнення
Захворювання крові небезпечні своїми наслідками, які не залежать від того, чи вчасно розпочато лікування чи ні. Патологічні процеси можуть сильно вплинути на працездатність людини, призвести до інвалідності або спровокувати смерть.
До найчастіших ускладнень відносяться:
- недокрів'я внаслідок сильної крововтрати;
- сепсис, що розвивається на тлі ослабленого імунітету після хіміотерапії або тривалого прийомумедикаментів;
- підвищена сприйнятливість до інфекційних та вірусних захворювань;
- розвиток супутніх хвороб внутрішніх органів (серця, судин, печінки, шлунка);
- поява геморагічного синдрому - схильність до крововтрат (розвивається на тлі занедбаної анемії).

Хвороби крові негативно впливають на серцево-судинну систему
Тривале лікування патологій крові може перерости в хронічні форми перебігу хвороби. Такий стан важче піддається терапії та може сильно вплинути на якість життя пацієнта.
Профілактика хвороб крові
Не допустити розвитку серйозних захворюваньможна, якщо вести здоровий спосіб життя та намагатися уникати впливу негативних факторів.
- Вчасно звертатись до лікаря, якщо неодноразово спостерігається погіршення самопочуття без видимих причин.
- Не запускати глистні інвазії та захворювання інфекційної природи.
- Стежити за харчуванням – регулярно приймати вітаміни. Раціон має бути повноцінним, але без шкідливої їжі.
- У побуті мінімізувати використання хімічних речовин. Максимально обмежити контакт із лакофарбовими виробами, бензолом та важкими металами.
- Уникати стресів та емоційних переживань. Більше відпочивати, нормалізувати сон.

Щоб уникнути хвороб крові, вживайте здорову їжу.
З метою профілактики хвороб крові та для зміцнення організму рекомендується більше часу проводити на свіжому повітрі, займатися помірними фізичними навантаженнями, уникати переохолодження чи перегріву.
Патологічні зміни в крові та кровотворних органах підступні. Серйозні хвороби можуть вражати всі вікові категорії людей, бути спадковими або набутими внаслідок супутніх порушень в організмі, а також мати доброякісний та злоякісний характер. Щоб не допустити важких ускладненьважливо не ігнорувати симптоматику, а вчасно звертатися до лікаря.
11-02-2012, 19:47
Опис
Анемії
Анемією, або недокрів'ям, називають стан, який характеризується зменшенням кількості еритроцитів та зниженням вмісту гемоглобіну в одиниці об'єму крові. У ряді випадків при анемії виявляються якісні зміни еритроцитів.
При анемії внаслідок порушення транспортної функціїрозвиваються явища гіпоксії, ознаками якої є задишка, тахікардія, неприємні відчуттяв області серця, запаморочення, слабкість, швидка стомлюваність, блідість шкірних покривів та видимих слизових оболонок. Виразність зазначених симптомів залежить від ступеня анемії та швидкості її розвитку. При глибокій анемії поряд із зазначеними симптомами виникають і порушення зору.
За колірним показникоманемії ділять на гіпохромні, нормохромні та гіперхромні. За величиною середнього діаметра еритроцитів анемії поділяють на мікроцитарні, нормоцитарні та макроцитарні. За характером регенерації розрізняють анемії регенераторні, гіпорегенераторні, гіпо- та апластичні, диспластичні або дизеритропоетичні.
В даний час загальноприйнятою класифікацією, побудованою за патогенетичним принципом з урахуванням етіологічних та найважливіших клініко-морфологічних форм, є класифікація, запропонована Г. А. Алексєєвим (1970).
I. Анеміївнаслідок крововтрат (постгеморагічні).
ІІ. Анеміївнаслідок порушеного кровообігу:
A. Залізодефіцитні анемії (хлоранемії).
Б. Залізонасичені, сидероахрестичні анемії.
B. В12 (фолієво)-дефіцитні, «перніціозні» анемії:
1. Екзогенна недостатність вітаміну В12 (фолієвої кислоти).
2. Ендогенна недостатність вітаміну B12 (фолієвої кислоти):
а) порушена асиміляція харчового вітамінуВ12 внаслідок випадання секреції шлункового мукопротеїну;
б) порушена асиміляція вітаміну B12 (фолієвої кислоти) у кишечнику;
в) підвищена витрата вітаміну B12 (фолієвої кислоти).
Г. В12 (фолієво)-«ахрестичні» анемії.
Д. Гіпоапластичні анемії:
1. Внаслідок дії екзогенних факторів.
2. Внаслідок ендогенної аплазії кісткового мозку.
Е. Метапластичні анемії.
ІІІ. Анеміївнаслідок підвищеного кроворуйнування (гемолітичні):
А. Анемії, зумовлені екзоеритроцитарними гемолітичними факторами.
Б. Анемії, зумовлені ендоеритроцитарними факторами:
1. Еритроцитопатії.
2. Ензімопенії:
а) дефіцит глюкозо-6-фосфат-дегідрогенази;
б) дефіцит піруваткінази;
в) дефіцит глутатіон-редуктази.
3. Гемоглобінопатія.
Нижче описуються характерні особливості окремих форм анемій, у яких найчастіше зустрічаються очні симптоми.
Гостра постгеморагічна анеміярозвивається внаслідок гострої одноразової та повторної крововтрати від травм, кровотеч із шлунково-кишкового тракту, при позаматкової вагітності, маткових кровотечах та ін. Симптоми хвороби патогенетично пов'язані зі зменшенням маси циркулюючої крові та кисневою недостатністю Клінічна картина в перші моменти після масивної крововтрати укладається в клініку пост-геморагічного шоку або колапсу: блідість шкірних покривів, непритомний стан, запаморочення, холодний піт, Частий ниткоподібний пульс, іноді блювання, судоми. Надалі в міру поліпшення загального стану та стабілізації артеріального тискуу клінічній картині починають переважати симптоми недокрів'я та гіпоксії. Саме цьому періоді найчастіше виявляються ознаки порушення зору до повного амаврозу, оскільки специфічні елементи сітківки дуже чутливі до анемії.
При хронічних гіпохромних залізодефіцитних анеміях, включаючи ранній і пізній хлороз, симптоматичних залізодефіцитних анеміях а, глистова, спруанемія , целіакія та ін.) Виразність очних симптомів залежить від ступеня анемії, яка, щоправда, індивідуально варіює в широких межах. Особливо часто зміни на очному дні виникають при концентрації гемоглобіну нижче 5 г% та рідше 7 г%.
Очне днопри анемії виглядає блідим. Цей симптом не завжди можна оцінити через відмінності в пігментації сітківки та хоріоїди. Легше виявляється деколорація диска зорового нерва та судин сітківки. При цьому артеріальні судинимають тенденцію розширюватися і наближатися калібром до аналогічних венозних гілочкам. Множинні крововиливиу сітківку – найбільш характерний симптом ретинопатії при анеміях (рис. 34).

Мал. 34.Очне дно при перніціозної анемії.
Причина крововиливу не цілком зрозуміла. Очевидно, нестача киснюобумовлює підвищену проникність капілярів. При перніціозної анемії має значення і супутня тромбоцитопенія.
Смугасті або у формі полум'я геморагії розташованіу шарі нервових волокон. Вони можуть локалізуватися в будь-якому відділі сітківки, але їх немає у жовтій плямі. Тому гострота зору зазвичай зберігається. Іноді у екстравазатах видно білий центр. Цей симптом найчастіше спостерігається при перніціозній анемії. У деяких випадках ішемія може бути причиною набряку диска зорового нерва та прилеглої сітківки. Зазвичай набряк виражений нерізко, але описані випадки застійного диска. Крім набряку у шарі нервових волокон можуть бути невеликі білі осередки, які складаються з фібрину і зазвичай добре розсмоктуються при покращенні стану хворого.
Значно важчі зміни сітківки спостерігаються при серповидноклітинної (дрепаноцитарної) анемії. Це захворювання відноситься до спадково-сімейної гемолітичної анемії, характерною особливістю якої є властивість еритроцитів набувати серповидної форми – цим захворюванням страждають переважно негри та рідко – особи білої раси. У Радянському Союзі описані поодинокі випадки.
Захворювання відноситься до групи гемоглобінопатійз уродженою неповноцінністю еритроцитів, зокрема з наявністю у них патологічного глобуліну.
Захворювання проявляється вже у дитячому віці та характеризується хронічною течієюз частими загостреннями у вигляді гемолітичних арегенераторних, тромботичних та секвестральних кризів.
При гемолітичних кризахвміст еритроцитів може протягом короткого періоду знижуватися до 1-2 млн., в 1 мм3 крові. Криз супроводжується розвитком жовтяниці та абдомінального синдрому. Арегенераторні кризи – це тимчасове, функціональне виснаження кістково-мозкового кровотворення. Тромботичні або болючі кризи, які іноді домінують у проявах хвороби, виникають на грунті генералізованих тромбозів дрібних судин, особливо черевної порожнини та кінцівок. Секвестральні кризи - це стани, що нагадують шок із раптовим розвитком анемії без гемолізу [Токарев Ю. Н., 1966].
Як і при інших уроджених гемолітичних анеміях, хворі серповидноклітинною анемією інфантильні, страждають на гіпогонадизм, мають баштовий череп та ін. При цьому захворюванні: особливо чітко виражений кістково-суглобовий синдром (дактиліти, болі, деформації, некрози суглобових головок і кісток). На гомілках часто розвиваються хронічні виразки. Селезінка та печінка збільшені. Тромбози та емболії – дуже характерна ознака. Поразки сітківки локалізуються переважно в екваторіальній та периферичній зонах та проходять 5 стадій. І стадія характеризується периферичною артеріолярною обструкцією, ІІ – появою артеріовенозних анастомозів. У III стадії розвивається неоваскулярна та фіброзна проліферація, яка призводить у IV стадії до крововиливів у склоподібне тіло. Зрештою (V стадія) розвивається відшарування сітківки.
Лейкози
Під лейкозами розуміють неопластичні захворювання, Пухлинна маса яких складається з клітин крові або, що, мабуть, більш точно, з клітин, подібних за своїм виглядом з клітинами крові.
Деякими вченими пухлини крові поділяютьсяна гемобластоми та гематосаркоми на тій підставі, що в окремих випадках кістковий мозок може бути повсюдно заселений цими пухлинними клітинами, а в інших випадках їхнє розростання здійснюється екстрамедулярно. На наш погляд, такий підрозділ часто здійснити дуже складно, тому що пухлинні розростання лейкемічних клітин можуть мати екстрамедулярну локалізацію і у хворих, у яких захворювання почалося з ураження кісткового мозку. І, навпаки, часом гематосарком згодом може відбуватися залучення у процес кісткового мозку, і клініцисти змушені у випадках говорити про лейкемизации процесу. На наш погляд, правильнішим є всі пухлини кровотворної тканини об'єднати під назвою «лейкози», оскільки пухлинна неопластична природа цих захворювань, що підкреслюється в назвах «гемобластози» або «гематосаркоматози», ні в кого практично не викликає сумнівів.
Етіологія лейкозівне може вважатися остаточно з'ясованою, що стосується, втім, інших пухлин однаковою мірою. Однак в даний час можна вважати встановленим, що такі фактори, як вірус, іонізуюча радіація, певні хімічні субстанції, у тому числі деякі лікарські речовини типу левоміцетину, бутадіону і цитостатиків, здатні надавати певний стимулюючий вплив на виникнення цих захворювань. Про роль спадкових чинників у виникненні лейкозів є цілком обгрунтовані думки. Вони підтверджуються випадками виникнення однотипних лейкозів у однояйцевих близнюків, великої схильності до розвитку лейкозів у хворих зі спадковими порушеннями генетичного апарату - хворобою Дауна, синдромами Тернера,.. Клайнфелтера і т. п. При цьому зазначено, що певні види лейкозів генетичні порушення. Необхідно мати на увазі, що сучасні наукові дані дуже переконливі на користь припущення про походження всієї лейкемічної маси з однієї мутованої клітини, що вийшла з-під контролу організму хворого. Такими є наявність кільцевої хромосоми в пухлинних клітинах хворих на гострі лейкози, що розвинулися у осіб, лікованих радіоактивним фосфором, різке збільшення вмісту однотипного за фізико-хімічними властивостями протеїну у хворих на парапротеїнемічні гемобластози. Філадельфійська хромосома у хворих на хронічний мієлолейкоз.
У клінічній практиціЛейкози прийнято підрозділяти залежно від типу клітини, що становить основу пухлинної маси. Ті лейкози, які протікають із проліферацією клітин малодиференційованих і не здатних до подальшої диференціації, зазвичай без лікування дуже злоякісні та називаються гострими. Лейкози, пухлинну масу яких складають клітини, що диференціюються і зрілі, зазвичай мають відносно доброякісний перебіг і звуться хронічних лейкозів.
Гострі та хронічні лейкозисвоєю чергою поділяються залежно від цього, яка клітина становить субстрат пухлини. В даний час описані лейкози, що розвиваються з клітин всіх паростків кровотворення - еритроїдного, тромбоцитарного. гранулоцитарного та агранулоцитарного типу. При цьому розрізняють гострі лейкози мієло-, моно-, мегакаріо-, еритро- та плазмобластного типів. Так як диференціація гострих лейкозів здійснюється лише на підставі цитохімічних методів дослідження, а цитохімічні способи ідентифікації клітин здійснюються за допомогою емпірично відібраного набору методів, з'явилися повідомлення про існування такої форми гострого лейкозу як недиференційований. Походження останнього, мабуть, можна віднести за рахунок проліферації клітин, що походять з більш ранніх, недиференційованих клітин кровотворення. Серед хронічних лейкозів виділені та продовжують виділятися форми лейкозів, в основі яких лежить проліферація будь-якої зрілої клітини крові. Тут і хронічний лімфолейкоз, хронічний мієлолейкоз, хронічний моноцитарний лейкоз, хронічний мегакаріоцитарний лейкоз, еритромієлоз, еритремія, плазмоцитома, хронічний базофільноклітинний лейкоз; є повідомлення про наявність хронічного еозинофільного лейкозу.
При сучасному рівні медичної науки, що дозволяє розрізняти найтонші деталі клітин, виробляються підрозділи в рамках ніби давно усталених форм лейкозів. Так, серед групи хворих з хронічним лімфолейкозомв даний час вже виділяють групи осіб, які страждають від проліферації як Т-, так і В-лімфоцитів, а серед хворих хронічним мієлолейкозомрозрізняють групи з проліферацією клітин, що мають філадельфійську хромосому та не мають її. Не виключена можливість, що ідентифікація лейкозів буде продовжуватися і надалі, і це дозволить здійснювати більш специфічне та дієве лікування хворих.
На підставі викладеного вище говорити про діагностику як самого лейкозу, так і про його конкретну форму досить нескладно. Діагностика цього захворюванняздійснюється при констатації гіперплазії кровотворної тканини, яка може мати місце як у периферичній крові, так і в кістковому мозку. При цьому в окремих осіб гіперплазія лейкемічних клітин має місце лише в кістковому мозку, а в периферичній крові ці клітини з'являються лише на пізніших стадіях захворювання. У зв'язку з цим дослідження кістковомозкового кровотворення за допомогою аналізу даних стернального пунктату, а іноді й структури кісткової тканиниза допомогою трепанобіопсії повинні проводитися в процесі діагностики. Застосування цитохімічних і цитогенетичних методів дослідження зазвичай призводить лише до уточнення варіанту лейкозу.
Можливість існування лейкемоїдних реакцій, Т. е. таких розростань кровотворної тканини, що виникають у відповідь на наявність в організмі хворого якогось фактора, що активує кровотворення, змушує іноді проводити спеціальні дослідження, що виключають наявність цих причин гіперплазії кровотворної тканини.
клінічна картиналейкозів дуже різноманітна. У цьому різноманітність клінічних проявів є в хворого як гострими, і хронічними лейкозами. Передбачити подальший клінічний перебіг, клінічні прояви лейкозу в окремого хворого, мабуть, не наважиться жоден досвідчений клініцист. Зробити це практично неможливо у зв'язку з тим, що висока морфодинамічність і майже повсюдне можливе поширення лейкемічної тканини в організмі хворого можуть демонструвати різноманітну симптоматику, симулюючи, особливо в початкових стадіях, захворювання різного характеру Приклад цього може бути робота одного з основоположників вітчизняної гематології акад. І. А. Кассирського, який разом зі своїми співробітниками при аналізі первинних діагнозів, з якими хворі надходили до клініки та у яких згодом був верифікований гострий лейкоз, виявив понад 60 різних нозологічних форм, серед яких фігурували сепсис, рак шлунка, ревматизм та гостра кишкова непрохідність, інфаркт міокарда, ревматоїдний артрит, гострий менінгіт та багато інших захворювань.
В той же час про клініку лейкозів говорити можна і досить просто у зв'язку з тим, що всі клінічні прояви цих захворювань можна об'єднати і зрозуміти на підставі розпізнавання головних синдромів, які зазвичай з тією чи іншою перевагою залежно від типу лейкозу мають місце у клінічній картині захворювання. Серед цих синдромівнайчастіше зустрічаються такі: 1) загальнотоксичний синдром (або інтоксикаційний); проявом його є лихоманка, слабкість, пітливість, схуднення, відсутність апетиту тощо; 2) геморагічний синдром. Прояви його надзвичайно різноманітні, включаючи менорагії, шкірні крововиливи та крововиливи в головний мозок; 3) синдром токсико-некротичних уражень слизових оболонок шлунково-кишкового тракту; 4) анемічний синдром; 5) синдром пухлинного росту, що характеризується розростанням лейкемічної тканини в організмі. Сюди ж потрібно відносити збільшення лімфатичних вузлів, печінки, селезінки, порушення функцій внутрішніх органів за рахунок їх здавлювання або порушення цілісності лейкемічною тканиною, що розростається.
Крім проявів цих синдромів, характерних для всіх лейкозів, окремі види лейкозів, зокрема парапротеїнемічні гемобластози(плазмоцитома, хвороба Вальденштрема, хвороби важких та легких ланцюгів), еритремія, мають у клінічній картині ряд особливостей, які будуть описані в окремих розділах Особливого забарвлення клінічної картини лейкозів (лімфатичного типу) іноді можуть надавати аутоімунні реакції , що виявляються гемолітичною анемією, лихоманкою, шкірними змінами тощо.
Не зупиняючись на зовнішніх проявах кожного з вище перерахованих синдромів, хотілося б відзначити, що за останні роки в клінічній картині лейкозів стали відзначатись прояви, які можуть бути пояснені як цитостатичною терапією, і подовженням терміну життя хворих із цією патологією. До них відносяться почастішання інфекційних ускладнень, які є причиною смерті майже 40% хворих на хронічний лімфолейкоз, почастішання неврологічної симптоматики (особливо у хворих на гострі лейкози, що отримали назву нейролех коза), а також часте розвиток у хворих на лейкози сечокислої нефропатії.
Таким чином, клініка лейкозів може характеризуватися найрізноманітнішою симптоматикоюяка є наслідком різноманітного поєднання зазначених вище синдромів. Звичайно, при окремих видах лейкозів можна відзначити переважання того чи іншого синдрому з перерахованих вище, проте недооцінювати можливості підключення будь-якого з них у клінічній карті при будь-якому вигляді лейкозу не може жоден клініцист.
Говорячи про лейкози, не можна не згадати того великого прогресу, який досягнуто сучасною медициною в лікуванні цих захворювань. Адже саме при цьому виді пухлин отримано результати, що дозволяють говорити про принципове лікування людини від злоякісного неопластичного захворювання. Лікування хворих на гострий лімфобластний лейкоз, лімфогранулематоз дозволяє сподіватися, що ці успіхи поширяться і на лікування інших форм лейкозів.
Гострі та хронічні форми лейкозів супроводжуються однаковими очними проявами, викликаними підвищенням в'язкості крові, гіпоксією та лейкемічною інфільтрацією тканини Ці зміни включають утворення мікроаневризм у судинах сітківки, геморагії, клітинну інфільтрацію хоріоїди, сітківки, зорового нерва і періорбітальних структур. Інфільтрація оболонок мозку може призвести до паралічу екстраокулярних м'язів та розвитку застійного диска. Описано також інфільтрацію повік, кон'юнктиви, орбітальної клітковини з розвитком екзофтальму.
При офтальмоскопії відзначається блідий фон очного дна. Ретинальні вени розширені, звивисті, і в сітківці по їх ходу нерідко видно білуваті смуги, що являють собою периваскулярну лейкемічну інфільтрацію. Артерії змінені значно менше, ніж вени.
Величина та форма геморагій варіює. Вони можуть бути глибокими, поверхневими чи навіть преретинальними. Нерідко в центрі ретинального крововиливу можна бачити білу ділянку, спричинену акумуляцією лейкоцитів. У найважчих випадках з'являються ішемічні ватоподібні вогнища у шарі нервових волокон, виражений набряк диска зорового нерва та перипапілярної сітківки та новоутворені ретинальні судини.
Зміни на очному дніпри лейкозах зустрічаються приблизно 70% випадків, особливо часто при гострих формах. Тяжкість змін більш менш корелює з тяжкістю хвороби, і при ефективному лікуванні основного захворювання покращується і стан очного дна.
Поліцитемії
Термін «поліцитемія» включає групу захворювань, які проявляються збільшенням маси еритроцитів в організмі, тобто збільшенням їхнього обсягу на 1 кг маси тіла. Число еритроцитів у 1 мм3 крові при поліцитемії підвищується до 7-10 млн., а вміст гемоглобіну до 180-240 г/л. Розрізняють «справжню» поліцитемію (еритремія, хвороба Вакеза) та вторинні (симптоматичні) еритроцитози.
Еритремія- первинне мієлопроліферативне захворювання кровотворної системи, в основі якого лежить тотальна гіперплазія клітинних елементів кісткового мозку, особливо його зорового паростка. Тому підвищений вміст лейкоцитів (до 9000-15 000 млн. в 1 мм3 крові) та тромбоцитів (до 1 млн. і більше) у крові поряд з помітнішим збільшенням кількості еритроцитів є досить характерною ознакою еритреміі. G. F. Stroebe (1951) виділив три гематологічні варіанти еритреміі: 1) без істотного збільшення числа лейкоцитів та Змін у формулі крові; 2) з помірним лейкоцитозом, нейтрофплезом та паличкоядерним зрушенням; 3) з високим лейкоцитозом, нейтрофілозом та зрушенням у формулі крові до мієлоцитів. При «справжній» поліцитемії виявляються ознаки мієло фіброзу та остеомієлосклерозу з мієлоїдною метаплазією селезінки. Як і при інших мієлопроліферативних захворюваннях, у сироватці крові хворих на поліцитемію нерідко виявляється підвищення концентрації лужної фосфатази, сечової кислоти та вітаміну В12. клінічна картина істинної поліцитеміїрізна залежно від фази хвороби та тяжкості перебігу.
У розгорнутій, власне еритремічній фазі хвороби характерними симптомами є: 1) зміна забарвлення шкірних покривів та видимих слизових оболонок; 2) збільшення селезінки та печінки; 3) підвищення артеріального тиску; 4) тромбози та геморагії.
Шкірні покриви зміненоу переважної більшості хворих. Вони набувають червоно-ціанотичного відтінку. Особливо чітко змінюється забарвлення щік, кінчиків вух, губ та долонь. Наголошуємо, що у фарбуванні шкірних покривів переважає червоний тон, але не яскравий, а вишневий. Подібний відтінок набувають і видимі слизові оболонки губ, язика та м'якого піднебіння. Судини склер помітно ін'єктовані (симптом кролячих очей). На щоках, губах, кінчику носа, особливо у жінок, нерідко виявляються телеангіектазії.
Дуже характерним симптомом еритремія є спленомегаліящо пов'язано з мієломною її метаплазією та підвищеним кровонаповненням. У хворих на справжню поліцитемію зазвичай збільшена та печінка. Збільшення її розмірів пов'язують також із підвищеним кровонаповненням, мієлоїдною метаплазією, розростанням сполучної тканини аж до розвитку цирозу або тромбозу внутрішньопечінкових вен (синдром Бадда-Кіарі). У ряду хворих перебіг хвороби ускладнюється розвитком холелітіазу та хронічного холецистогепатиту. До розвитку цих ускладнень призводить властива хворим на еритремію пленохромія жовчі.
Майже у половини хворих на еритреміювиявляється гіпертонія, патогенез якої розглядається у плані компенсаторної реакції організму у відповідь на зменшення ударного та хвилинного об'єму крові, підвищення її в'язкості та збільшення периферичного опору(А. В. Демідова, Є. М. Щербак). Поєднання підвищеного артеріального тиску із збільшеною селезінкою є кардинальною ознакою істинної поліцитемії. Якщо при цьому у хворого збільшується маса еритроцитів, діагноз поліцитемії стає безперечним.
Характерна парадоксальна схильність хворих до поліцитеміїі до тромбозів (великих артеріальних та венозних судинмозку, серця, печінки та селезінки, дрібних судин кистей та стоп) та до підвищеної кровоточивості (з виразок шлунка та дванадцятипалої кишки, після екстракції зубів, шкірні геморагії та кровотечі зі слизових оболонок). Причиною кровоточивості при істинній поліцитемії є збільшення маси циркулюючої крові з переповненням нею судин та паретичним розширенням капілярів, а також дефіцит плазмових факторів згортання, зокрема фібриногену [Мачабелі М. С., 1962], серотоніну [Матвеєнко JI. А., 1965].
Розвиток тромбозів при еритремііпов'язано з підвищенням в'язкості крові, уповільненням струму крові, збільшенням числа тромбоцитів та еритроцитів, зі склеротичним ураженням стінок судин, загальною гіперкоагулабільністю крові.
У хворих на еритремію нерідко уражаються нирки (у них розвиваються інфаркти внаслідок тромбозу судин або нирковокам'яної хвороби як результат порушення пуринового обміну, що властиво мієлопроліферативним захворюванням).
Справжня поліцитемія характеризується тривалою течією , яке може бути легким, середньої тяжкостіта важким. У розвитку хвороби виділяють три періоди, чи фази. Перша фаза захворювання протягом тривалого періоду може протікати латентно або з нерезко вираженими клінічними симптомами. На ранніх стадіях захворювання нерідко приймається за гіпертонічну хворобу.
Описана клінічна картина характеризує розгорнуту другу, так звану еритремічну фазу. І в цій фазі перебіг захворювання може бути різноманітним.
Термінальна фаза характеризується розвитком вторинного лшелофіброзу з анемією та зникненням зовнішніх ознак еритремії або розвитком гострого гемоцитобластозу, рідше – ретикульозу.
На відміну від істинної поліцитемії вторинні еритроцитоз є не самостійними нозологічними одиницями, а лише симптомами інших захворювань. Збільшення кількості еритроцитів та гемоглобіну пов'язане не з проліферативним процесом у кістковому мозку, а з його функціональним подразненням (абсолютні еритроцитози) або зі згущенням крові без посилення еритропоезу (відносні еритроцитози). У наведеній нижче класифікації вказані основні види вторинних еритроцитозів, варіанти їх перебігу, основні патогенетичні механізми, що лежать в основі їх розвитку, та конкретні захворювання, що супроводжуються розвитком вторинних еритроцитозів.


Симптом поліцитемії, що найбільш кидається в очі - плетора обличчя та кон'юнктиви. Кон'юнктивальні та епісклеральні судини, особливо вени, розширені, звивисті, насичено-червоного кольору. Такий самий вигляд мають судини сітківки (рис. 35).

Мал. 35.Очне дно за поліцитемії.
Привертає увагу темно-червоний колір очного дна. Диск зорового нерва також надзвичайно червоного кольору. Нерідко можна бачити більш менш виражений набряк диска зорового нерва і перипапілярної сітківки і поодинокі геморагії.
У деяких випадках розвивається оклюзія центральної вени сітківки. Оклюзія, мабуть, має неповний характер. Прогноз у разі зазвичай сприятливий, у разі значно кращий, ніж за оклюзії центральної вени сітківки інший етіології.
Парапротеїнемії
До цієї групи захворювань належать передусім мієломна хвороба(плазмоклітинний парапротеїнемічний ретикульоз або хвороба Рустицького) та макроглобуліновий ретикулолімфоматоз(Хвороба Вальденстрема, або макроглобулінемічна пурпура).
Мієломна хворобає системним захворюванням крові пухлинно-гіперпластичного типу із злоякісною проліферацією клітин ретикулоплазматичного типу. Це - лейкоз-ретикулоз, зокрема плазмоклітинний пара-(або пато-) протеїнемічний ретикульоз.
Залежно від переважаючого виду клітин виділяють три типи мієлом: 1) ретикулоплазмоцитому, 2) плазмобластому та 3) плазмоцитому.
Протеїнурія- Дуже частий симптом мієломної хвороби. З сечею виділяється, зазвичай, мікромолекулярний білок (білок Бенс-Джонса). З протеїнурією пов'язано розвиток мієломної нефропатії - парапротеїнемічного нефрозу, який зазвичай закінчується смертю при явищах азотемічної уремії.
З високою концентрацією білка в крові пов'язана і властива мієломна хвороба висока в'язкість крові.
Хвороба Вальденстремав даний час розглядається як макроглобуліновий ретикулолімфоматоз, характерною особливістю якого є здатність синтезувати макроглобуліни: у крові з'являються глобуліни з молекулярною масою понад 1 ТОВ ТОВ. Захворюють переважно люди похилого віку. У клінічній практиці переважає геморагічний синдром, іноді з надзвичайно рясними носовими кровотечами. Механізм геморагічного синдрому складний і до кінця не встановлений. Припускають, що він пов'язаний, з одного боку, з неповноцінністю тромбоцитів, що взаємодіють з макроглобулінами, а з іншого - з підвищеною проникністю стінок судин внаслідок інфільтрації їх патологічними білками, високої в'язкості крові та внутрішньосудинної аглютинації еритроцитів.
Виділяють переважноскелетні форми та скелетно-вісцеральні форми захворювання. У патогенетичному плані клінічна картина хвороби зводиться до двох синдромів, а саме до ураження кісток та патології білків крові. Поразка кісток проявляється болями, переломами та розвитком пухлин. Особливо часто уражаються хребет, кістки таза, ребра та череп з розвитком відповідної неврологічної симптоматики.
Вісцеральна патологія проявляєтьсяголовним чином ураженням печінки, селезінки, лімфатичних вузлів та нирок. Її розвиток пов'язаний як із специфічною клітинною інфільтрацією зазначених органів, так і з вираженими змінами білків крові, з накопиченням у крові аномального білка – парапротеїну, що продукується мієломними клітинами. При мієломній хворобі протеїнемія може сягати 12-18 г%.
Ретинопатіяпри початкових формахмієломної хвороби та хвороби Вальденстрема відсутня. У ряду хворих очне дно є своєрідною картиною fundus paraproteinemicus. Характерно розширення ретинальних вен та збільшення їх звивистості. Артерії також розширюються, але значно меншою мірою. Потім проявляються симптом перехреста (передавлювання вени під артерією), мікроаневризми, оклюзія дрібних вен, геморагії у сітківку. У деяких випадках виникають також ватоподібні вогнища у шарі нервових волокон сітківки та набряк диска зорового нерва.
Вважають, що зазначені зміни у сітківці пов'язаніяк з гіперпарапротеїнемією, так і з високою в'язкістю крові. В азотемічній стадії хвороби розвивається характерна для хронічних захворювань нирок ретинопатія.
Що стосується змін судин сітківки, то їх зв'язок з підвищеною в'язкістю плазми крові був продемонстрований експериментально. Після введення мавпам у кров декстрану з високою відносною масою на очному дні були виявлені розширені та звивисті ретинальні судини, особливо вени, мікроаневризми та геморагії.
Мієломна хвороба може вражатитакож кістки орбіти, повіки, слізну залозу, слізний мішок та кон'юнктиву, інфільтрувати склеру, райдужку, хоріоїдею, сітківку та зоровий нерв. Ці поразки, однак, не пов'язані з підвищеною в'язкістю крові.
Геморагічні діатези
Під геморагічні діатези маються на увазі такі патологічні стани, які проявляються п підвищеною кровоточивістюза відсутності істотних ушкоджень судинної стінки, т. е. кровотечі розвиваються у ситуаціях, коли в інших здорових у плані людей їх немає.
Важливість проблемигеморагічних діатезів дуже велика. По-перше, це пояснюється тим, що кількість людей, які страждають на підвищену кровоточивість, у світі перевищила шестизначну цифру. По-друге, люди, які страждають на геморагічні діатези, не можуть вважатися повноцінними членами суспільства, оскільки їх потенційні можливості різко обмежені як анемізацією, часто супутньої цієї патології, так і тими видами діяльності, які оберігають судини хворого від різних пошкоджень.
По-третє, важливість інформації про наявність у хворих на геморагічний діатез визначається тим, що багато форм цього страждання протікають приховано або виявляються слабо, маючи моносимптомну клініку. У разі необхідності оперативних втручань, навіть таких незначних, як екстракція зуба або тонзилектомія, а також при призначенні деяких ліків, таких як ацетилсаліцилової кислоти, геморагічні діатези можуть загрожувати самому життю хворого.
Патогенез геморагічних діатезів нині можна вважати досить добре вивченим. Як відомо, обмеження кровоточивостіу здорової людини при пошкодженні судинної стінки здійснюється за рахунок наступних механізмів: скорочення судини у місці її пошкодження, осідання на місці пошкодження судини циркулюючих тромбоцитів та утворення ними первинної гемостатичної пробки та закріплення її фібриновою стінкою з формуванням остаточної «вторинної» гемостатичної проб. Порушення будь-якого з цих механізмів призводить до порушення процесу гемостазу та розвитку геморагічного діатезу.
Сучасні уявлення про механізми зсідання крові дозволяють нам запропонувати наступну робочу класифікацію геморагічних діатезів.
КЛАСИФІКАЦІЯ ГЕМОРРАГІЧНИХ ДІАТЕЗІВ
I. Геморагічні діатезиобумовлені дефектом прокоагулянтів (гемофілії):
а) недостатня кількість одного або кількох факторів, що беруть участь у формуванні фібрину;
б) недостатня активність факторів-прокоагулянтів;
в) наявність інгібіторів окремих прокоагулянтів у крові хворого.
ІІ. Геморагічні діатезиобумовлені дефектом тромбоцитарної ланки гемостазу:
а) недостатня кількість тромбоцитів (тромбоцитопенії);
б) функціональна неповноцінність тромбоцитів (тромбоцитопатії);
в) поєднання кількісної та якісної патології тромбоцитів.
ІІІ. Геморагічні діатези, що виявляються в результаті надлишкового фбрінолізу:
а) ендогенного;
б) екзогенного.
IV. Геморагічні діатези, що проявляються в результаті патології судинної стінки:
а) уроджені;
б) набуті.
V. Геморагічні діатези, що розвиваються внаслідок поєднання кількох причин (тромбогеморагічний синдром, хвороба Віллебранда).
Найчастішою причиноюгеморагічних діатезів є дефект тромбоцитарної ланки гемостазу, що є причиною кровоточивості у 80% хворих [Марквардт Ф., 1976]. У групі хворих на геморагічні діатези, що розвиваються при неповноцінності прокоагулянтної ланки гемостазу, найчастіше діагностується гемофілія А (65-80%), гемофілія В (13-18%) і гемофілія С (1,4-9%).
Історично ж склалося так, що більшої популярності набули геморагічні діатези, зумовлені дефектом формування фібрину. В даний час відомо, що утворення фібрину забезпечується шляхом правильної взаємодії білків прокоагулянтів, більшість з яких має власний номер, який вказує римська цифра. Розрізняють 13 субстанцій, серед яких фібриноген (фактор I), протромбін (II), проакцелерин-акцелерин (V), проконвертин (VII), антигемофілічний глобулін А (VIII), крістмас-фактор (IX), фактор Стюарта – Прауера (X) , Попередник тромбопластину плазми (XI), фактор Хагемана (XII), фібринстабілізуючий фактор (XIII) Крім них, три виявлені останнім часом фактори не мають цифрового позначення. Це фактори Флетчера, Фіцжеральця та Пасової.
Кількісний або якісний дефект будь-якого із зазначених вище прокоагулянтів, а також поява в крові хворого на інгібітор цього фактора здатні викликати геморагічний стан у хворого.
Велика кількість цих станів, яка підходить до цифри 30, а також велика схожість їх клінічних проявів дозволяють об'єднувати дані захворювання під загальною назвою. гемофілії».
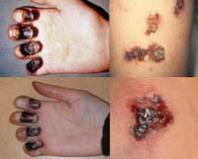
Для гемофілій характернівеликі, глибокі, зазвичай ізольовані, спонтанні синці та гематоми, часті крововиливи в суглоби при вкрай рідкісному розвитку шкірних і слизових «пурпур» у рідкісних і слабко виражених кровотечах при поверхневих пошкодженнях шкіри. Грубі лабораторні пробидемонструють подовження часу зсідання крові за відсутності порушення часу кровотечі. Практичні лікарі повинні чітко уявляти, що точна діагностика причини геморагічного діатезу можлива лише при застосуванні спеціальних. лабораторних методівдослідження, без чого адекватна терапія практично неможлива.
Серед геморагічних діатезів, що розвиваються при неповноцінності тромбоцитарної ланки гемостазу, найчастіше зустрічаються ті, що обумовлені зменшенням кількості тромбоцитіву кровотоку хворого. Ці стани, які називають синдромом Верльгофа, неоднорідні з своєї причини. Кількість тромбоцитів може знижуватися як внаслідок утворення проти них аутоантитіл (аутоімунні тромбоцитопенії), так і внаслідок неповноцінного утворення їх у кістковому мозку. Можливі також неповноцінність тромбоцитарної оболонки та їх цитоліз.
Увага клініцистів за останні роки виявилася прикутою до таких геморагічних станів; які зумовлені функціональною неповноцінністю тромбоцитів, не здатних забезпечувати повноцінний гемостаз навіть за достатньої їх кількості у кровотоку хворого. Після того, як така патологія вперше була описана Глянцманом, виявлено велику кількість патологічних форм, які обумовлені порушенням того чи іншого етапу формування тромбоцитарної пробки, що здійснюється тромбоцитами: їх адгезією, агрегацією, активацією прокоагулянтної ланки, ретракцією кров'яного згустку.
Виявлення цих дефектів, Виявлення їх поєднань з деякими іншими проявами захворювання призвело до опису низки окремих нозологічних форм. У той же час вивчення функцій тромбоцитів при низці описаних захворювань дозволило відзначити відсутність зв'язку між порушеннями тромбоцитарних функцій та іншими симптомами, які не стосуються гемостазу.
Різні поєднання дефектів тромбоцитарних функцій дозволили говорити про наявність цілої групи тромбоцитопатій, що виявляються найрізноманітнішими сполуками порушень таких функцій тромбоцитів, як адгезія, агрегація, реакція звільнення, активація прокоагулянтів, ретракція. При уточненні причини геморагічного діатезу необхідно детальне вивчення як кількісного, і якісного стану тромбоцитів за умов лабораторії.
У клінічній картині цих захворювань характерними є часті тривалі кровотечіпри поверхневих пошкодженнях шкіри, часті шкірні та слизові «пурпури», тоді як крововилив у суглоби, спонтанні синці та гематоми досить рідкісні.
Дефекти гемостазу, Викликані патологією судинної стінки, діагностуються досить легко в тих випадках, коли ця патологія доступна візуальному спостереженню: при хворобі Рандю-Ослера, синдромі Елерса-Данлоса, хвороби Гіппеля-Ліндау, синдромі Казабаха-Мерріта та ін. В даний час є вказівки, геморагічні діатези можуть розвиватися при неповноцінності колагену судинної стінки та порушеної внаслідок цього адгезії тромбоцитів. Однак цю патологію можна діагностувати лише при застосуванні складних лабораторних методів.
Велику увагу клініцистів останнім часом привертають випадки розвитку геморагійу хворих при множинному мікротромбуванні капілярів внутрішніх органів. Ці стани отримали назви тромбогеморагічного синдрому. Патогенез його пояснюється тим, що при масивному швидкому тромбоутворенні в згустку споживаються багато факторів згортання крові, особливо тромбоцити та фібриноген. Крім того, гіпоксія судинної стінки призводить до викиду в кровотік великої кількості активаторів плазміногену та підвищення фібринолітичної активності крові. Діагностика цих станів дуже важлива, оскільки потребує «парадоксального» застосування антикоагулянтів на лікування геморагій.
Цікаві знахідки виявлені щодо вивчення патогенезу кровоточивостіу пацієнтів із хворобою Віллебрандта, яка характеризується поєднанням симптомів, що відображають порушення як прокоагулянтної, так і тромбоцитарної ланок гемостазу. При цьому було виявлено, що антиген фактора VIII має суттєве значеннядля запуску адгезії тромбоцитів до пошкодженої поверхні та показав важливість взаємовідносин цих провідних механізмів зупинки кровотеч.
Велике різноманіття причин геморагічних діатезів, створення специфічних методів лікування цих станів зобов'язують практичних лікарівдо детального вивчення питань діагностики та лікування хворих із підвищеною кровоточивістю.
Найчастішими очними проявами при пурпурі є підшкірні та кон'юнктивальні геморагії. Крововиливи в сітківку дуже рідкісні. У тих випадках, коли вони все ж таки є, геморагії розташовуються в шарі нервових волокон. Слід мати на увазі, що при травмі ока, у тому числі хірургічної, можлива сильна кровотеча, особливо при гемофілії.
Розпізнавання проводиться на підставі даних гістологічного дослідженнябіопсії м'язів гомілки або передньої черевної стінки.
Лікування. Великі дози глюкокортикоїдних гормонів, цитостатичні препарати (циклофосфан, азатіоприн). При хронічному перебігу хвороби лікувальна фізкультура, масаж, водолікування, прийом делагілу, плаквеніла.
Розділ 6 Хвороби системи кровотворення
Анемія (малокровність). Зменшення у крові загальної кількості гемоглобіну. Найчастіше знижується і рівень еритроцитів. Анемії завжди вторинні, тобто одна із ознак якогось загального захворювання.
Анемії залізодефіцптис пов'язані з дефіцитом заліза в організмі. Це веде спочатку до множинних трофічних порушень (сухість шкіри, ламкість нігтів, випадання волосся), оскільки погіршується функція тканинних дихальних ферментів, що містять залізо, а потім порушується утворення гемоглобіну, розвивається. гіпохромна анемія(З низьким колірним показником). Організм дорослої людини втрачає залізо головним чином при хронічних крововтратах, не відновлюючи повною мірою цей елемент з їжею, у дітей подібні явища обумовлені малим вихідним надходженням до кровотворної системи плода через нестачу його у матері.
Симптоми та перебіг. Характерні млявість, підвищена стомлюваність, запор, головний біль, спотворення смаку (хворі їдять крейду, глину, з'являється схильність до гострої, солоної їжі тощо), ламкість, викривлення та поперечна смугастість нігтів, випадання волосся. Відзначаються і типові для всіх анемій ознаки, що відображають ступінь недокрів'я: блідість шкіри та слизових оболонок, серцебиття, задишка при фізичного навантаження. Важливий характер захворювання, що спричинив дефіцит заліза (виразка шлунка, 12-палої кишки, геморой, міома матки, рясні менструальні кровотечі).
Розпізнавання ґрунтується на виявленні змін в аналізах крові: зниження рівня гемоглобіну та еритроцитів, колірний показник нижче 0,8, змінені розміри та форма еритроцитів (анізоцитоз, пойкілоцитоз). Значно знижено вміст заліза у сироватці крові, її загальна залізозв'язувальна здатність, білка, що переносить залізо (феритин).
Лікування. Усунути причину крововтрат. Протягом тривалого періоду (кілька місяців і більше) призначають препарати заліза переважно всередину. Переливання крові не показано за винятком важких станів, пов'язаних із масивною крововтратою.
Гемолітичні анемії пов'язані з посиленим руйнуванням еритроцитів та збільшенням у крові вмісту продуктів їхнього розпаду - білірубіну, вільного гемоглобіну, або появою гемосидерину в сечі. Важлива ознака – значне наростання відсотка “новонароджених” еритроцитів – ретикулоцитів у результаті підвищеного утворення клітин червоної крові. Виділяють: а) анемії з переважно позасудинним (внутрішньоклітинним) гемолізом (розпадом) еритроцитів, зумовленим їх генетично структурною та функціональною неповноцінністю; б) анемії з внутрішньосудинним гемолізом, зазвичай з гострим руйнуванням еритроцитів при різних токсичних впливах, переливанні групопесумісної крові, холодова (при дії вкрай низьких температур), маршева (у солдатів після тривалих і виснажливих марш-кидків). Поділяються також на: 1) Вроджені гемолітичні анемії. До них відноситься група (сфероцитарна, ововоклітинна) зі спадковою аномалією оболонки еритроцитів, що веде до зміни їх форми і є причиною передчасного руйнування; інша група - зі спадковим дефіцитом різних ферментних систем еритроцитів, що сприяє їхньому швидшому руйнуванню; третя група - гемоглобіпопатій (серповидноклітинна, таласемія), при яких порушена структура або синтез гемоглобіну; 2) Придбані аутоімунні гемолітичні та ізоімунні анемії, зумовлені механічним ушкодженнямеритроцитів, а також токсичні мембранопатії.
Симптоми та перебіг Прояви залежать від форми гемолітичної анемії. При внутрішньоклітинному розпаді еритроцитів з'являється жовтяниця, збільшується селезінка, знижується рівень гемоглобіну, відзначається схильність до утворення каменів у жовчному міхурі, зростає кількість ретикулоцитів. При внутрішньосудинному гемолізі на додаток до цих ознак з'являються тромбози, можуть бути асептичні некрози трубчастих кісток, розвиваються виразки гомілок, при гемолітичному кризі виділяється темна сеча. При вроджених гемолітичних анеміях трапляється деформація лицевого черепа.
Розпізнавання проводиться на основі виявлення клінічних та лабораторних ознакгемолізу. З метою з'ясування його природи беруть проби Кумбса та Хема, цукрозну, визначають рівень сироваткового заліза, проводиться генетичне обстеження.
Лікування. Відміна медикаменту, що викликав гемолітичний криз (при дефіциті глюкозо-6-фосфатдегідрогенази), при гемолітичних кризах - інфузійна терапія, сечогінні, вітаміни, переливання еритроцитарної маси (відмиті еритроцити), у тяжких випадках - видалення глюкокортикоїди (предпізолон), імунодепресанти.
В12- та фолієводефіцитні анемії характеризуються порушенням синтезу ДНК та РНК у клітинах, званих мегалобластами, що призводить до повернення ембріонального типу кровотворення. Зустрічаються переважно вулиць похилого віку, можуть бути обумовлені як недостатнім надходженням в організм вітаміну В12 і фолієвої кислоти, так і недостатнім засвоєнням при різних захворюваннях шлунка, тонкого кишечниката печінки, при зараженні глистами. Одна з причин дефіциту вітаміну В12 – хронічна алкогольна інтоксикація.
Симптоми та перебіг. Уражаються кровотворна тканина, травна система ("полірована" мова, відчуття печіння в ньому, пригнічення шлункової секреції) та нервова система(Слабкість, стомлюваність, фунікулярний мієлоз). Відзначається невелика жовтяниця, у крові – підвищення непрямого білірубіну, збільшується селезінка, печінка.
Розпізнавання. У крові визначається анемія з колірним показником понад 1,0, мегалоцити, зниження кількості тромбоцитів та лейкоцитів, з'являються полісегментовані нейтрофіли. У кістковому мозку – переважання мегалобластів (при пункції кісткового мозку).
Лікування. Вітамін В12 у високих дозах, фолієва кислота. При нормалізації складу крові – тривала підтримуюча терапія цими препаратами.
Гіпопластичні та апластичні анемії характеризуються наростаючим зменшенням вмісту формених елементів (еритроцитів, лейкоцитів, тромбоцитів) у периферичній крові та кістковому мозку. Причиною може бути токсична дія деяких ліків, хімічних речовин, аутоагресія та поява антитіл до кровотворних клітин, іноді причини неясні (ідіопатична форма).
Симптоми та перебіг. Наростаюча анемія, зниження тромбоцитів та лейкоцитів, що може призводити до інфекційним ускладненням, підвищеної кровоточивості
Розпізнавання. Виявляється анемія із нормальним колірним показником. Вирішальною є картина кісткового мозку при стернальній пункції та трепанобіопсії - різке зменшення кількості клітин, заповнення кістковомозкового простору жиром.
Лікування. Глюкокортикоїдні гормони, анаболічні стероїди, видалення селезінки, пересадка кісткового мозку.
Геморагічні діатези. Характеризуються схильністю до кровоточивості. Розрізняють сімейні, або спадкові форми: вроджені аномалії тромбоцитів, дефіцит або дефект факторів згортання плазми, неповноцінність дрібних кровоносних судин. Набуті форми: синдром дисемінованого внутрішньосудинного згортання, імунні ураження судинної стінки та тромбоцитів, порушення нормального утворення клітин крові, токсико-геморагічне ураження кровоносних судин при геморагічних лихоманках, висипний тиф. Викликають також захворювання печінки, васкуліти, прийом антикоагулянтів, дезагрегантів, фібринолітиків, дефіцит вітаміну С.
Гемофілія. Спадкове захворювання, яким страждають лише чоловіки, хоча носіями дефектного гена є жінки. Порушення згортання обумовлено недоліком низки плазмових факторів, що утворюють активний тромбопластин. Найчастіше відсутній антигемофілічний глобулін. Захворювання проявляється у дитинстві тривалими кровотечами при незначних ушкодженнях. Можуть виникати носові кровотечі, гематурія – кров у сечі, великі крововиливи, гемартрози – кров у порожнині суглоба. Основні ознаки: подовження часу згортання, скорочення протромбінового часу.
Лікування – переливання свіжої крові чи плазми, введення спеціальної антигемофілічної плазми.
Ідіопатична тромбоцитопечична пурпура (хвороба Верльгофа). Характеризується кровоточивістю внаслідок зниження кількості тромбоцитів. Причина хвороби найчастіше імунна. Хвороба протікає хвилеподібно. Поза загостренням число тромбоцитів може бути нормальним або трохи зниженим. При зменшенні числа тромбоцитів нижче 40х10/л розвивається підвищена кровоточивість до виражених кровотеч, найчастіше носових, шлунково-кишкових, маткових, ниркових. На шкірі з'являється геморагічна висипка самостійно або після накладання джгута на руку - т.п. позитивні симптоми "щипка чи джгута". Селезінка збільшена. Аналіз крові показує збільшення часу кровотечі.
Лікування в період загострення – переливання тромбоцитарної маси, свіжої крові, застосування глюкокортикоїдних гормонів (Преднізолон), іноді видалення селезінки.
Спадкова геморагічна телсачгіектазія (хвороба Рандю-Ослера). Характеризується розвитком множинних легко кровоточивих розширених судин (телеангіоектазій), розташованих на різних ділянкахшкіри та слизових оболонках. Іноді перший і єдиний симптом – носові або шлунково-кишкові кровотечі. Вони виникають при незначному пошкодженні або самостійно і при частому повторенні призводять до розвитку залізодефіцитної анемії. Захворювання може ускладнитися цирозом печінки.
Розпізнавання засноване на виявленні типових телеаш-іоектазій рецидивуючих кровотечах з них, сімейному характері захворювання.
Лікування – зупинка кровотечі, при необхідності переливання крові, лікування залізодефіцитної анемії.
Геморагічний васкуліт (капіляротоксикоз, хвороба Шенлейн-Геноха). В основі хвороби – аутоімунне ураження ендотелію дрібних судин. Найчастіше з'являються дрібні геморагічні висипання, переважно на передній поверхні гомілок та стегон. Можуть бути біль у суглобах, артрити. У деяких випадках на перший план виступає ураження судин черевної порожнини з різкими болями у животі, шлунково-кишковими кровотечами. Хвороба протікає тривалий час, іноді з багаторічними ремісіями. Прогноз визначається ураженням нирок.
Лікування. Обмеження фізичного навантаження, при загостренні – постільний режим, антигістамінні та протизапальні препарати, у тяжких випадках призначають гепарин, глюкокортикоїдні гормони (преднізолон), амінохінолінові препарати (делагіл, плаквеніл), аскорбінову кислоту, рутин. Деяким хворим із хронічною рецидивною течією можна рекомендувати санаторно-курортне лікування (південь України, Південний берегКриму, Північний Кавказ).
Лейкози. Численні пухлини, що виникають з кровотворних клітин і вражають кістковий мозок За ступенем злоякісності виділяють гострі та хронічні лейкози. У групі хронічних найчастіше зустрічається мієло- та лімфолейкоз, а також мієломна хвороба, еритремія, остеомієлофіброз.
Гострий лейкоз - швидко прогресуюче захворювання, у якому відбувається зростання молодих недиференційованих клітин крові, втратили здатність до дозрівання. Виділяють 2 варіанти гострого лейкозу – гострий мієлобластний та гострий лімфобластний лейкоз, останній зустрічається частіше у дітей.
Симптоми та перебіг. Захворювання зазвичай супроводжується високою температурою слабкістю, розвитком тяжких кровотеч чи інших геморагічних проявів. Рано можуть приєднуватись різні інфекційні ускладнення, виразковий стоматит, некротична ангіна. Виникають болі в кінцівках, постукування по грудині та довгим трубчастим кісткам болюче. Може спостерігатися збільшення розмірів печінки, селезінки. Лімфовузли змінюються мало. У крові значно підвищується кількість молодих патологічних форм, про бластних клітин - лімфобластів, проміжні форми дозріваючих лейкоцитів відсутні. Загальна кількість лейкоцитів може бути збільшена або навіть зменшена.
Лікування – поєднання кількох цитостатичних препаратів, великих доз глюкокортикоїдних гормонів, лікування інфекційних ускладнень.
Хронічний мчелоленкоз характеризується порушенням нормального дозрівання гранулоцитарних лейкоцитів, появою вогнищ позакостномозкового кровотворення. Хвороба може протікати тривалий час з великими періодами ремісії після курсів лікування.
Симптоми та перебіг. Хворі скаржаться на підвищену стомлюваність, слабкість, поганий апетит, зниження ваги. Збільшується селезінка, печінка, можливі геморагічні прояви. У крові значно збільшується кількість лейкоцитів, анемія. Нерідко підвищується рівень сечової кислоти у сироватці крові. На пізній стадії хвороби знижується кількість тромбоцитів, виникають інфекційні ускладнення, схильність до тромбозів, в аналізі крові виявляються мієлобласти, мієлоцити.
Розпізнавання проводиться на підставі даних дослідження кісткового мозку (стернальна пункція, трепанобіопсія).
Лікування. У термінальному періоді хвороби (бластний криз) лікування проводиться як за гострого лейкозу. Поза загостренням - підтримуюче лікування мієлосаном, мієлобромолом.
Хронічний ламфолечкоз. Патологічне розростання лімфоїдної тканиниу кістковому мозку, лімфовузлах, селезінці, печінці, рідше в інших органах. Хвороба виникає у похилому віці та протікає тривало.
Симптоми та перебіг. Зниження ваги, слабкість, втома, втрата апетиту виражені не різко. Відзначається збільшення різних груп лімфовузлів у всіх областях тіла: шийних, пахових, стегнових, надключичних, ліктьових. Вони щільні, безболісні, рухливі. Рентгенографія виявляє збільшені вузли в коренях легень. Іноді вони здавлюють трахею, стравохід, порожні вени. Селезінка та печінка також збільшуються. У крові підвищується число лейкоцитів переважно за рахунок лімфоцитів, серед них зустрічаються лімфоцити, що розпадаються (клітини Боткіна-Гумпрехта), відзначається анемія і тромбоцитопенія (зниження числа тромбоцитів).
Розпізнавання проводиться поданим дослідженням кісткового мозку.
Лікування у легких випадках не проводиться. При здавленні лімфовузлами сусідніх органів – рентгенотерапія. При швидкому розвитку хвороби призначають глюкокортикоїдні гормони, цитостатики.
Лімфогранулематоз – хронічне прогресуюче захворювання, пухлина лімфатичних вузлів з наявністю клітин Березовського-Штернберга. Причина невідома.
Симптоми та перебіг. Іноді хвороба починається із проявів інтоксикації (висока температура, слабкість, пітливість), підвищується ШОЕ, збільшуються лімфовузли. Вони щільні, еластичні, частіше не спаяні між собою. У разі їх некротичного розпаду з'являються нориці. Часто буває свербіж шкіри. Зрідка відзначається первинна локалізація лімфогранулематозу в шлунку, легені, селезінці. У крові знижується число лімфоцитів, підвищується кількість нейтрофілів з помірним паличкоядерним зрушенням, підвищена ШОЕ.
Розпізнавання – на підставі характерних гістологічних ознак хвороби у лімфовузлі, взятому за біопсії.
Лікування. Курси поліхіміотерапії, що чергуються курсами рентгенотерапії.
Розділ 7 Хвороби вітамінної недостатності
Група захворювань, викликаних недостатністю в організмі одного або багатьох вітамінів, розвиваються зазвичай або за нестачі їх у їжі, або за порушення їх всмоктування зі шлунково-кишкового тракту.
Недостатність вітаміну А. Вітамін А міститься у багатьох продуктах тваринного походження (вершкове масло, яєчний жовток, печінка риб), у рослинних продуктах міститься каротин (провітамін А). Вітамін А жиророзчинний. Добова потреба для дорослої людини 15 мг (5000 ME).
Симптоми та перебіг. У першу чергу змінюються органи зору: характерна нічна, або "куряча", сліпота через дистрофічні зміни сітківки і зорових нервів, виникає сухість кон'юнктиви, на ній утворюються прозорі бляшки, покривається виразками рогівка. Відбуваються дистрофічні зміни шкіри, слизових та шкірних залоз: сухість, лущення та блідість шкіри, атрофія потових та сальних залоз, схильність до гнійничкових захворювань. Можливі інфекційні ураженнясистеми дихання, сечовиділення, травлення, загальне нездужання, слабкість, у дітей – затримка росту та розвитку, порушення з боку нервової системи.
Розпізнавання проводиться на основі даних анамнезу та клінічної картинипри біохімічному дослідженні сироватки крові рівень вітаміну А знижується нижче 100 мкг/л, каротину - нижче 200 мкг/л.
Лікування. Повноцінне харчуванняіз включенням продуктів, багатих ретинолом та каротином: м'ясна, молочна їжа, морква, томати, абрикоси. Можливе призначення ретинолу у добовій дозі дорослому від 10000 до 100000 МО/добу протягом 2-4 тижнів. Великі дози та безконтрольне лікування можуть спричинити гіпервітаміноз А.
Недостатність вітаміну B1 (тіаміну). До гіповітамінозу та авітамінозу B1 (бері-бері) привертають вагітність, лактація, важка фізична праця, гарячкові захворювання, тиреотоксикоз, цукровий діабет. Добова потреба у вітаміні становить близько 2 мг.
Симптоми та перебіг. Поширена поразка периферичних нервів(поліневрит) та серцево-судинної системи. Хворі скаржаться на стомлюваність, біль голови, задишку, серцебиття при фізичному навантаженні. Турбують парестезії (відчуття "повзання мурашок") та зниження шкірної чутливості ніг, а потім та інших ділянок тіла, відчуття слабкості та тяжкості в ногах, кульгавість, набряки, прискорений пульс, зниження артеріального тиску. Можливі розлади функцій органів травлення, зору, психіки.
Розпізнавання проводиться на підставі даних клінічної картини, рівень тіаміну в сечі нижче 100 мкг, вміст кокарбоксилази в еритроцитах нижче 20-40 мкг/л.
Лікування. Призначають вітамін B1 у комплексі з іншими вітамінами (нікотинова кислота, рибофлавін, вітамін В6), проводять симптоматичну терапію. Вживають їжу з високим змістомвітаміну B1 пшеничний та житній хліб, дріжджі, горох, жовток яєць, свинину.
Недостатність вітаміну В2 (рибофлавіну). Міститься у багатьох продуктах тваринного та рослинного походження.
Бере участь у здійсненні зорової функції ока та синтезі гемоглобіну. Добова потреба дорослої людини становить 2-3 мг.
Симптомії протягом. Зниження апетиту, схуднення, слабкість, головний біль, порушення сутінкового зору, дистрофічні зміни шкіри та слизових оболонок: відчуття печіння шкіри, різь в очах, поява кон'юнктивіту, тріщин і скоринок у кутах рота, афтозного стоматиту, глоситу (язика яскраво-червона) ), себорейного дерматиту, особливо вираженого в області носогубної складки, крилах носа, вухах, сухого дерматиту, що свербить на кистях рук. При тривалому перебігу спостерігаються зміни нервової системи, анемія.
Перебіг хвороби хронічний із загостреннями у весняно-літні місяці.
Розпізнавання ґрунтується на характерних клінічних ознаках; вміст рибофлавіну у сироватці крові нижче 3 мкг/л.
Лікування. Повноцінне харчування (яйця, молочні продукти, дріжджі, хліб, крупи), прийом рибофлавіну (у складі комплексних вітамінних препаратів).
Недостатність нікотинової кислоти (вітамін ВЗ, РР). Тяжкий ступінь недостатності проявляється симптомами пелагри. Добова потреба дорослих у нікотиновій кислоті становить 20-25 мг.
Симптоми та перебіг. Уражаються переважно травна, нервова система та шкіра. Показовими є втрата апетиту, сухість і печіння в роті, блювання, пронос, що чергується із запором, загальна прогресуюча слабкість. Мовно-червоний, набряковий з хворобливими виразками, пізніше "лаковий".
Далі:
Система крові включає:
- органи та тканини кровотворення, або гемопоезу, в яких дозрівають формені елементи крові;
- периферичну кров, яка включає циркулюючу та депоновану в органах та тканинах фракції;
- органи кроворуйнування;
Система крові є внутрішнім середовищем організму та однією з його інтегруючих систем. Кров виконує численні функції - дихання, обмін речовин, екскреції, терморегуляції, підтримки водно-електролітного балансу. Вона здійснює захисні та регуляторні функції завдяки наявності в ній фагоцитів, різних антитіл, біологічно активних речовин, гормонів. На процеси кровотворення впливають багато чинників. Важливе значеннямають особливі речовини, що регулюють проліферацію та дозрівання клітин крові, - гемопоетини, Проте загальний регулюючий вплив має нервова система. Усі численні функції крові спрямовано підтримку гомеостазу.
Картина периферичної крові та кісткового мозку дозволяє судити про функції багатьох систем організму. При цьому найбільш повне уявлення про стан кровотворної системи можна отримати, лише досліджуючи кістковий мозок. Для цього спеціальною голкою (трепаном) проводять пункцію грудини чи гребеня клубової кісткиі одержують кістковомозкову тканину, яку потім досліджують під мікроскопом.
Морфологія кровотворення
Усі формені елементи крові в нормальних умовахутворюються в червоному кістковому мозку плоских кісток – грудини, ребер, кісток таза, хребців. У трубчастих кістках дорослої людини кістковий мозок представлений переважно жировою тканиною і має жовтий колір. Діти в трубчастих кістках відбувається кровотворення, тому кістковий мозок червоний.
Морфогенез кровотворення.
Родоначальником всіх клітин крові є стовбурова кровотворна клітина кісткового мозку, яка трансформується в клітини-попередники, які морфологічно не відрізняються один від одного, але дають початок мієло- і лім-фопоез (рис. 42). Ці процеси регулюються гемопоетинами, серед яких виділяють еритро-, лейко- та тромбоцитопоетини. Залежно від переважання тих чи інших поетинів посилюється мієлопоез і клітини-попередники починають трансформуватися у бластні форми мієлоцитарного, еритроцитарного та тромбоцитарного паростків крові. При стимуляції лімфопоезу починається дозрівання лімфоцитарного, а також моноцитарного паростків крові. Таким чином відбувається розвиток зрілих клітинних форм – Т- та В-лімфоцитів, моноцитів, базофілів, еозинофілів, нейтрофілів, еритроцитів та тромбоцитів.
на різних етапахгемопоезу внаслідок патологічних впливів можуть виникати порушення дозрівання кровотворних клітин та розвиваються хвороби крові. Крім того, на багато патологічних процесів, що виникають в організмі, система крові реагує зміною свого клітинного складу та інших параметрів.
Порушення обсягу циркулюючої крові
Мал. 42. Схема кровотворення (за І. Л. Чорткову та А. І. Воробйову).
При різних хворобах та патологічних процесахможе змінюватися загальний об'єм крові, а також співвідношення її формених елементів та плазми. Виділяють 2 основні групи порушень об'єму крові:
- гіперволемії - стани, що характеризуються збільшенням загального обсягу крові та. зазвичай, зміною гематокриту;
- гіповолемії - стани, що характеризуються зменшенням загального обсягу крові та поєднується зі зниженням або збільшенням гематокриту.
ГІПЕРВОЛЕМІЇ
Види:
- Нормоцитемічна гіперволемія - стан, що проявляється еквівалентним збільшенням обсягу формених елементів та рідкої частини циркулюючої крові. Гематокрит залишається у межах норми. Такий стан виникає, наприклад. при переливанні великої кількості (щонайменше 2 л) крові.
- Олігоцитемічна гіперволемія - стан, що характеризується збільшенням загального обсягу крові внаслідок зростання переважно обсягу плазми. Показник гематокриту при цьому нижчий за норму. Така гіперволемія з'являється при введенні великої кількості фізіологічного розчину або кровозамінників, а також при недостатності функції виділення нирок.
- Поліцитемічна гіперволемія - стан, що проявляється збільшенням загального обсягу крові внаслідок переважного підвищення числа її формених елементів, насамперед еритроцитів. При цьому гематокрит стає вищим за норму. Найчастіше таке явище спостерігається при тривалій гіпоксії, що стимулює вихід еритроцитів з кісткового мозку в кров, наприклад, у жителів високогір'я, на певних етапах патогенезу низки захворювань легень та серця.
ГІПОВОЛЕМІЇ
Види:
- Нормоцитемічна гіповолемія - стан, що проявляється зменшенням загального обсягу крові за збереження гематокриту у межах норми, що спостерігається відразу після крововтрати.
- Олігоцитемічна гіповолемія характеризується зменшенням загального обсягу крові з переважним зниженням кількості її формених елементів. Гематокрит при цьому нижче за норму. Спостерігається також після крововтрати, але в пізніші терміни, коли з міжклітинного простору до судин надходить тканинна рідина. У цьому випадку об'єм циркулюючої крові починає зростати, а кількість еритроцитів залишається низьким.
- Поліцитемічна гіповолемія - стан, у якому зниження загального обсягу крові обумовлено переважно зменшенням обсягу плазми. Показник гематокриту при цьому вищий за норму. Таке згущення крові спостерігається при втраті рідини після великих опіків, при гіпертермії з масивним потовиділенням, холері, що характеризується неприборканим блюванням і проносом. Згущення крові сприяє утворенню тромбів, а зменшення загального обсягу крові нерідко призводить до серцевої недостатності.
ПАТОЛОГІЯ СИСТЕМИ ЕРИТРОЦИТІВ
Анемія, або недокрів'я, - зниження загальної кількості гемоглобіну в організмі та, як правило, гематокриту. У більшості випадків анемії супроводжуються еритропенією - зниженням кількості еритроцитів в одиниці об'єму крові нижче норми (менше 3109/л у жінок і 4109/л у чоловіків). Винятком є залозодефіцитна анеміята таласемія, при яких кількість еритроцитів може бути нормальною або навіть збільшеною.
Значення анемії для організму визначається насамперед зменшенням кисневої ємності крові та розвитком гіпоксії, з якою пов'язані основні симптоми розладів життєдіяльності цих хворих.
Види анемій:
- внаслідок крововтрати – постгеморагічні;
- внаслідок порушеного кровотворення – дефіцитні;
- внаслідок підвищеного кроворуйнування – гемолітичні.
За течією анемії можуть бути гострими та хронічними.
За змінами структури еритроцитів при анеміях виділяють:
- анізоцитоз, що характеризується різною формою еритроцитів;
- пойкілоцитоз – характеризується різними розмірами еритроцитів.
При анеміях змінюється кольоровий показник - Вміст гемоглобіну в еритроцитах, який в нормі дорівнює I. При анеміях він може бути:
- більше 1 (гіперхромна анемія);
- менше 1 (гіпохромна анемія).
АНЕМІЇ СЛІДСТВО КРОВОВТРАТИ (ПОСТГЕМОРРАГІЧНІ)
Ці анемії завжди вторинні, оскільки виникають унаслідок хвороб чи поранень.
Гостра постгеморагічна анемія виникає при гострій крововтраті. наприклад із судин дна виразки шлунка, при розриві маткової трубиу разі трубної вагітності, з легеневих каверн при туберкульозі тощо (внутрішня кровотеча) або з пошкоджених судин при пораненнях кінцівок, шиї та інших частин тіла (зовнішня кровотеча).
Механізми розвитку гострих постгеморагічних станів. На початковому етапі крововтрати більшою чи меншою мірою знижується обсяг циркулюючої крові та розвивається гіповолемія. У зв'язку з цим зменшується приплив венозної кровідо серця. його ударний та хвилинний викид. Це зумовлює падіння рівня артеріального тиску та ослаблення серцевої діяльності. В результаті зменшується транспорт кисню та субстратів метаболізму з крові до клітин, а від останніх – вуглекислого газу та відпрацьованих продуктів обміну речовин. Розвивається гіпоксія, яка багато в чому визначає результат крововтрати. Останній ступінь зазначених розладів в організмі позначається як постгеморатичний шок.
Морфологія
Проявами гострої анемії є блідість шкірних покривів та недокрів'я внутрішніх органів. У зв'язку з різким зменшенням оксигенації тканин підвищується вироблення еритропоетину, що стимулює еритропоез. У кістковому мозку при цьому відбувається значне збільшення числа клітин еритроїдного ряду і кістковий мозок набуває малинового кольору. У селезінці, лімфатичних вузлах, периваскулярній тканині з'являються осередки позакостномозкового, або екстрамедулярного, кровотворення. Нормалізація показників периферичної крові після поповнення крововтрати настає приблизно через 48-72 год.
Порушення гемодинаміки та зниження інтенсивності біологічного окислення в клітинах зумовлюють включення адаптивних механізмів :
- активацію тромбоутворення;
- реакції серцево-судинної компенсації крововтрати у вигляді звуження просвіту дрібних судин та викиду крові з депо;
- підвищення серцевого викиду;
- підтримання обсягу циркулюючої крові за рахунок надходження в судини рідини з інтерстицію.
Хронічна постгеморагічна анемія виникає при значній крововтраті внаслідок кровотеч, що повторюються, наприклад з гемороїдальних вен, при маткових кровотечах і т. п. Така крововтрата призводить до хронічної гіпоксії тканин і порушення в них обміну речовин.
Морфологія
Хронічна гіпоксія сприяє розвитку жирової дистрофії паренхіматозних органів. Жовтий кістковий мозок трансформується в червоний, тому що посилюються еритро- та мієлопоез. У печінці, селезінці, лімфатичних вузлах можуть з'являтися осередки екстрамедулярного кровотворення. Разом з тим при повторюваних і виражених ковопотерях може наступити гіпо- і аплазія кровотворної тканини, що вказує на виснаження гемопоезу.
АНЕМІЇ ВСЛІДСТВО ПОРУШЕНОЇ КРОВООСВІТИ (ДЕФІЦИТНІ)
Ці анемії є наслідком нестачі низки речовин, необхідні нормального гемопоэза - заліза, вітаміну B 12 , фолієвої кислоти та інших. Серед них найбільше значення має злоякісна анемія Аддісона-Бірмера. в основі якої лежить дефіцит вітаміну В 12 та фолієвої кислоти.
У 12-дефіцитна, або фолієводефіцитна, анемія. Етіологія анемії пов'язана з дефіцитом вітаміну В 12 та фолієвої кислоти, яка регулює нормальний гемопоез у кістковому мозку. Однак для активації фолієвої кислоти необхідно, щоб вітамін, що надходить з їжею, У 12 (зовнішній фактор)з'єднався з білком, що утворюється в шлунку - гастромукопротеїном (внутрішній фактор) , що виробляється додатковими клітинами залоз слизової оболонки шлунка Разом вони утворюють комплекс, який називається антианемічним фактором . Потім цей комплекс надходить у печінку та активує фолієву кислоту, а та у свою чергу стимулює еритропоез за еритробластічним типом. Якщо ж розвивається аутоімунний гастрит і з'являються антитіла до додаткових клітин або гастромукопротеїну, які знищують ці клітини або внутрішній фактор, то в слизовій оболонці шлунка не всмоктується вітамін 12 і не утворюється гастромукопротеїн. Така сама ситуація виникає при високій резекції шлунка з приводу пухлини або виразкового процесу.
Патогенез.
В результаті атрофії слизової оболонки шлунка аутоімунного характеру виникає дефіцит фолієвої кислоти та вітаміну В12. Порушується еритропоез і замість еритроцитів утворюються їхні попередники – великі мегалобласти, які з'являються у периферичній крові. Однак мегалобласти швидко руйнуються, розвиваються анемія та загальний гемосидероз. Крім того, при дефіциті вітаміну В 12 порушується утворення мієліну в оболонках нервових стовбурів, що порушує їхню функцію.
Патологічна анатомія.
У хворих відзначаються блідість шкірних покривів, водяниста кров, точкові крововиливи, через атрофію слизової оболонки язика він набуває малинового забарвлення ( гунтерівський глосит), характерні атрофічний гастрит, ущільнення та збільшення печінки через жирову дистрофію та гемосидероз, пов'язаних з гіпоксією та з посиленим руйнуванням мегалобластів. У спинному мозку- Розпад осьових циліндріву задніх та бічних стовпах та вогнища розм'якшення тканини мозку ( фунікулярний мієлоз), що супроводжується тяжкою неврологічною симптоматикою. Кістковий мозок плоских та трубчастих кісток червоний, нагадує малинове желе. У селезінці та лімфатичних вузлах осередки екстрамедулярного кровотворення.
Перебіг захворювання прогресує, з періодами ремісії та загострення. Лікування анемії препаратами фолієвої кислоти та вітаміну B 12 призвело до того, що хворі перестали помирати від цього захворювання.
АНЕМІЇ НАСЛІДКИ ПІДВИЩЕНОГО КРОВОРОЗРУШЕННЯ — ГЕМОЛІТИЧНІ
Для цих анемій характерне переважання процесу руйнування еритроцитів (гемолізу) над їх утворенням. Тривалість життя еритроцитів при цьому знижена і не перевищує 90-100 днів.
Види гемолітичних анемій
За походженням гемолітичні анемії ділять на набуті (вторинні) та вроджені чи спадкові.
Отримані гемолітичні анемії можуть бути викликані численними факторами. Етіологія цих анемій пов'язана з дією факторів фізичного, хімічного та біологічного, у тому числі аутоімунного характеру, особливо при дефіциті речовин, що стабілізують мембрани еритроцитів, наприклад а-токоферолу. Найбільше значення мають звані гемолітичні отрути хімічного (сполуки миш'яку, свинцю, фосфору та інших.) і біологічного походження. Серед останніх - отрути грибів, різні токсичні речовини, що утворюються в організмі при тяжких опіках, інфекційні хвороби (наприклад, малярія, зворотний тиф), переливання крові, несумісної за групою або резус-фактором.
Патогенез.
Гемоліз еритроцитів може відбуватися всередині судин та за їх межами. При цьому розпадається гемоглобін і з гему синтезуються два пігменти - гемосидерин та білірубін. Тому гемолітичні анемії зазвичай супроводжуються розвитком загального гемосидерозу та жовтяниці. Крім того, еритропенія та розпад гемоглобіну призводять до появи вираженої гіпоксії, що супроводжується жировою дистрофією паренхіматозних органів.
Морфологія гемолітичних анемій характеризується розвитком гіперпластичних процесів у кістковому мозку, у зв'язку з чим він набуває малинового кольору, появою вогнищ екстрамедулярного кровотворення, вираженою жовтяницею шкірних покривів та внутрішніх органів, гемосидерозом та жировою дистрофією печінки, серця та нирок.
Гемолітична хвороба новонароджених є прикладом набутих гемолітичних анемій та має велике значення в акушерській та педіатричній практиці. В її основі лежить імунний конфлікт між матір'ю і плодом по резус-фактору, який має антигенні властивості. Цей фактор вперше був виявлений в еритроцитах мавп макак резусів і має 80-85% людей. Якщо мати резус-негативна, тобто не має резус-фактора, а плід резус-позитивний, то в організмі матері утворюються антитіла проти еритроцитів плода і у нього виникає внутрішньосудинний гемоліз еритроцитів.

Мал. 43. Серповидно-клітинна анемія. Еритроцити серповидної форми. Електронограма.
При цьому плід може загинути на 5-7-му місяці вагітності, а у новонароджених розвивається гемолітична анемія, що супроводжується недокрів'ям та жировою дистрофією внутрішніх органів, вираженою жовтяницею та гемосидерозом.
Спадкові, або вроджені, гемолітичні анемії пов'язані з генетичним дефектом структури мембран, ферментів або гемоглобіну. Цей дефект передається у спадок.
Види: уроджені гемолітичні анемії залежно від генетичного дефекту можуть бути обумовлені мембранопатіями, ферментопатіями, гемоглобінопатіями.
Патогенез всіх вроджених гемолітичних анемій в основному подібний - внаслідок того чи іншого генетичного дефекту або руйнується мембрана еритроцитів, а самі еритроцити зменшуються в розмірах і можуть набувати сферичної форми ( мікросфероцитоз),або підвищується проникність мембрани та еритроцити збільшуються у розмірах за рахунок надходження надлишкової кількості рідини, або порушується синтез гемоглобіну ( гемоглобінози) і утворюються еритроцити неправильної форми, що містять гемоглобін, що швидко розпадається, причому утримує кисень (таласемія, серповидно-клітинна анемія та ін.)(Рис. 43).
Морфологія уроджених гемолітичних анемій мало відрізняється від змін при вторинних гемолітичних анеміях, за винятком розмірів та форми еритроцитів. Також характерні виражений внутрішньосудинний гемоліз, гіпоксія, гемосидероз, жирова дистрофія паренхіматозних органів, гіперплазія кровотворної тканини, можливі осередки екстрамедулярного кровотворення, гепато- та спленомегалія.
ПАТОЛОГІЯ СИСТЕМИ ЛЕЙКОЦИТІВ
У крові здорової людини в умовах спокою натще міститься 4109/л лейкоцитів. Багато лейкоцитів знаходиться у тканинах, де вони беруть участь у імунному контролі.
Типові зміни кількості лейкоцитів в одиниці об'єму крові характеризуються їх зниженням - лейкопенії, або збільшенням - лейкоцитози, що, як правило, є реакцією системи лейкоцитів, що розвивається при хворобах і патологічних станах. Тому лікування хвороби призводить до нормалізації лейкоцитарної формули.
Лейкопенія - зменшення кількості лейкоцитів в одиниці об'єму крові нижче за норму, зазвичай менше 4 10 9 /л. Вона виникає внаслідок пригнічення білого паростка системи гемопоезу, при посиленому руйнуванні лейкоцитів або при перерозподілі крові між кровоносним руслом та депо крові, що спостерігається, наприклад, при шоці.
Значення лейкопенії полягає в ослабленні захисних сил організму та підвищенні його сприйнятливості до різних інфекційних збудників.
Види лейкопеній за походженням:
- первинні лейкопенії(Вроджені або спадкові) пов'язані з різними генетичними дефектамиу системі кровотворення на різних етапах лейкопоезу;
- вторинні лейкопеніївиникають при дії на організм різних факторів – фізичних (іонізуючих випромінювань тощо), хімічних (бензол, інсектициди, цитостатики, сульфаніламіди, барбітурати та ін.), продуктів метаболізму або компонентів різних збудників хвороб.
Лейкоцитарна формула- Співвідношення різних видів циркулюючих лейкоцитів.
Якщо збільшується кількість молодих форм нейтрофілів (паличкоядерних, метамієлоцитів, мієлоцитів, промієлоцитів), розташованих у лівій частині лейкоцитарної формули, говорять про зсув формули вліво, що вказує на посилення проліферації клітин мієлоцитарного ряду. У правій частині формули розташовуються зрілі форми цих клітин. Лікування хвороби призводить до нормалізації лейкоцитарної формули. Зменшення нормального числа лейкоцитів у лейкоцитарній формулі вказує на зниження регенераторних можливостей мієлоїдної тканини.
Патогенез лейкопеній відображає порушення або пригнічення процесу лейкопоезу, а також надмірне руйнування лейкоцитів у циркулюючій крові або в органах гемопоезу, перерозподіл лейкоцитів у судинному руслі, можлива також втрата лейкоцитів організмом. При цьому внаслідок пригнічення регенерації лейкопоетичної тканини початкових етапахлейкопенії знижується кількість молодих форм нейтрофілів, а збільшення їх молодих форм (тобто зсув лейкоцитарної формули вліво) вказує на припинення дії, що пошкоджує, і активацію лейкопоезу. Можлива також поява анізоцитозу та пойкілоцитозу лейкоцитів.
Лейкоцитоз- збільшення кількості лейкоцитів в одиниці об'єму крові вище 4109/л. Він може бути фізіологічним, адаптивним, патологічним або мати форму пейкемоїдної реакції.
- Фізіологічний лейкоцитоз виникає у здорових людейу зв'язку із перерозподілом крові під час травлення, при фізичній роботі.
- Адаптивний лейкоцитоз розвивається при захворюваннях, що особливо характеризуються запаленням. При цьому кількість лейкоцитів може збільшуватися до 40109/л.
- Патологічний лейкоцитоз відбиває пухлинну природу лейкоцитозу та характеризує лейкоз.
Лейкемоїдна реакція- Підвищення загального чиста лейкоцитів периферичної крові більше 40 10 9 /л з появою їх незрілих форм (промієлоцитів, мієлобластів), що робить лейкоцитоз схожим на лейкоз.
Види лейкоцитозу пов'язані зі збільшенням тих чи інших форм лейкоцитів:
Агранулоцитоз- Відсутність або значне зниження абсолютного числавсіх видів зернистих гранулоцитів (лейкоцитів) – нейтрофілів, еозинофілів, базофілів. Агранулоцитоз, як правило, поєднується з лейкопенією.
ПУХЛИНИ СИСТЕМИ КРОВІ, АБО ГЕМОБЛАСТОЗИ
Гемобластози - пухлинні захворювання кровотворної та лімфатичної тканини. Вони поділяються на системні захворювання - лейкози , та регіонарні - злоякісні лімфоми, або гематосаркоми . При лейкозах первинно уражається кістковий мозок і пухлинні клітини виявляються в крові (лейкемія), а при лімфомах у термінальній стадії настає широке метастазування зі вторинною поразкоюкісткового мозку. За поширеністю гемобластози займають 5 місце серед усіх пухлин людини. Діти перших 5 років життя їх частку припадає 30 % випадків онкологічних захворювань.
Етіологія гемобластом принципово не відрізняється від причин, що викликають інші пухлини (див. розділ 10) - це різні мутагенні фактори екзо-і ендогенного походження, що діють на стовбурові та напівстволові клітини-попередниці. Велике значенняу виникненні гемобластоз має спадковий фактор.
Патогенез.
Безліч етіологічних факторіввпливають на геном стовбурових та напівстволових клітин, призводячи до їх злоякісної трансформації. Тому геном є так званим вузьким місцем, через яке мутагени впливають на протоонкогени та антионкогени, перетворюючи їх на клітинні онкогени, що призводить до появи пухлини. Розвиток гемобластозів починається з малігнізації однієї стовбурової або напівстволової клітини, що дає пул. пухлинних клітин. Отже, всі гемобластози мають моноклонове походження, і всі наступні пухлинні клітини розвиваються з клітини, що спочатку мутувала, і відносяться до одного клону. Крім малігнізації на рівні стовбурових і напівстволових клітин-попередниць, розвивається ще блок диференціювання в пулі пухлинних клітин і вони втрачають здатність до дозрівання.
Лейкози
Лейкози- системні пухлинні захворювання, що виникають із кровотворних клітин з ураженням кісткового мозку.
Захворюваність на лейкози коливається від 3 до 10 на 100 000 населення. Чоловіки хворіють у 1,5 рази частіше за жінок. Гострі лейкози найчастіше спостерігаються у віці від 10 до 18 років, а хронічні – у людей віком від 40 років.
Морфогенез.
При лейкозах пухлинна тканина спочатку розростається на території кісткового мозку і поступово пригнічує та витісняє нормальні паростки кровотворення. Тому у хворих на лейкоз розвиваються анемія, тромбоцит-, лімфоцит-, гранулоцитопенія, що призводить до підвищеної кровоточивості, крововиливів, зниження імунітету і приєднанням інфекційних захворювань. Метастазування при лейкозах полягає у появі лейкозних інфільтратів у печінці, селезінці, лімфатичних вузлах, стінках судин та ін. Обтурація судин пухлинними клітинами призводить до розвитку інфарктів органів та виразково-некротичних ускладнень.
Класифікація лейкозівзаснована на 5 ознаках цих захворювань.
- За ступенем диференціювання пухлинних клітин
виділяють недиференційовані, владні та цитарні лейкози. При високому рівніблоку диференціювання клітини пухлини нагадують недиференційовані та бластні форми гемопоезу. Такі лейкози протікають гостро та дуже злоякісно.
При зупинці диференціювання на рівні процитарних та цитарних клітин-попередниць лейкози протікають хронічно та менш злоякісно. - За цитогенетичною ознакою гострі лейкози поділяють на лімфобластний, мієлобластний, монобластний, еритромієлобластний, мегакаріобластний, недиференційований. Хронічні лейкози ділять на лейкози мієлоцитарного походження (хронічний мієлоцитарний, хронічний нейтрофільний, хронічний еозинофільний та ін.), лімфоцитарного (хронічний лімфолейкоз і парапротеїнемічні лейкози - мієломна хвороба, первинна макроглоб. цитарний лейкоз, гістіоцитоз X.
- За імунним фенотипом пухлинних клітин: виявлено маркерів їх антигенів.
- За загальною кількістю лейкоцитів у периферичній крові
виділяють лейкози:
- лейкемічні- десятки та сотні тисяч лейкоцитів в 1 мкл крові, у тому числі бласти;
- сублейкемічні- Число лейкоцитів крові становить 25-50 10 9 /л, включаючи бластні форми;
- лейкопенічні- кількість лейкоцитів у периферичній крові нижча за норму, але є бласти;
- алейкемічні- кількість лейкоцитів у крові менше норми і відсутні бластні форми.
- За характером течії виділяють:
- гострі лейкози (вони ж недиференційовані та бластні);
- хронічні лейкози (цитарні).
Гострі лейкози розвиваються зі всіх паростків морфологічно недиференційованих кровотворних клітин-попередниць. Тривалість перебігу захворювання становить 2-18 місяців, при успішному лікуванні ремісії можуть тривати до 5-8 років.
Морфогенез.
Різні форми гострих лейкозо» мають стереотипні морфологічні прояви. Вони заклинаються у розвитку лейкозної інфільтрації кісткового мозку атиповими клітинами ранніх стадій гемопоезу (рис. 44). Зважаючи на нециференційованість цих клітин їх цитогенетичну приналежність можна виявити лише за допомогою цитохімічних та імуногістохімічних методів. Кістковий мозок трубчастих кісток стає червоним, при деяких гострих лейкозах він набуває зеленого кольору, властивого гною, - піоїдний кістковий мозок.При цьому відбувається витіснення нормальних клітингемопоезу пухлинними клітинами. У периферичній крові та в кістковому мозку є лише бластні та зрілі форми клітин, але відсутні їх проміжні форми. Така картина крові називається « лейкемічний провал «. Лейкозні інфільтрати виявляються у лімфатичних вузлах, селезінці та печінці, що призводить до збільшення запалення порожнини рота та тканини мигдаликів ускладнюється некротичним гінгівітом, тонзилітом, некротичною ангіною, а при інфільтрації оболонок мозку розвивається лейкозний менінг. Пригнічення еритроцитарного паростка призводить до наростаючої гіпоксії та жирової дистрофії паренхіматозних органів.

Мал. 44. Кістковий мозок при гострому лімфобластному лейкозі. Тканина мозку складається здебільшого з лімфобластів (а), просвіти судин заповнені тими ж клітинами (б).
Внаслідок тромбоцитопенії, ураження печінки та стінок судин у хворих розвивається геморагічний синдром аж до крововиливів у мозок та смертельних. шлунково-кишкових кровотеч. На цьому фоні іноді приєднується сепсис, що призводить до смерті (рис. 45).
Найчастіше, особливо у дітей, зустрічається гострий лімфобластний лейкоз,пов'язаний з пухлинною трансформацією попередників Т-і В-лімфоцитів, та гострий мієлобластний лейкоз,яким частіше страждають дорослі, зумовлений пухлинною проліферацією клітин-попередниць мієлоїдного ряду.

Мал. 45. Гострий лейкоз, а – лейкозна інфільтрація печінки (показано стрілками); б – некроз мигдалини (некротична ангіна); в - лейкозна інфільтрація нирок; г - множинні крововиливи в епікарді та ендокарді; д - лейкозна інфільтрація кісткового мозку (піоїдний кістковий мозок), стоншення кортикального шару стегнової кістки (показано стрілкою).

Мал. 46. Печінка при хронічному мієлолейкозі. Розростання клітин мієлоїдного роду (а) у процесі синусоїдів.
Хронічні лейкози течуть понад 4 роки, при успішному лікуванні ремісії захворювання можуть тривати 20 років і більше. Хронічні лейкози відрізняються від гострих цитарним диференціюванням пухлинних клітин і більш тривалим перебігом, який має певні стадії:
- моноклонова стадія характеризується присутністю лише одного клону пухлинних клітин, що протікає роками, щодо доброякісно;
- поліклонова стадія, або владний криз пов'язана з появою вторинних пухлинних клонів, характеризується швидким злоякісним перебігом, і 80% хворих гинуть саме в цій стадії.
Морфогенез.
Лейкозні інфільтрати розростаються в кістковому мозку, печінці, селезінці, нирках, лімфатичних вузлах, брижі кишечника, нерідко в середостінні, у зв'язку з чим ці органи і тканини різко збільшуються в розмірах і можуть здавлювати сусідні органи(Рис. 46). Особливо виражена спленомегалія (маса селезінки досягає 6-8 кг) та гепатомегалія (маса печінки 5-6 кг). У судинах утворюються лейкозні тромби, які можуть призвести до розвитку ішемічних інфарктів, частіше у селезінці та нирках. У крові наростає кількість нейтрофілів або лімфоцитів, багато перехідних клітинних форм. Виражена анемія, тромбоцитопенія, значна імунодепресія та схильність до інфекційних ускладнень, від яких хворі нерідко гинуть. Кістковий мозок сіро-червоний. Жирова дистрофія паренхіматозних органів надає їм сіро-жовте забарвлення.
Доброякісна течія змінюється бластним кризом. При цьому в крові швидко наростає кількість бластних форм - мієло-, еритро-, лімфо-, мегакаріобластів та ін. Загальна кількість лейкоцитів периферичної крові може досягати кілька мільйонів один мкл. Владний криз спричиняє смерть хворих.
ПАРАПРОТЕІНЕМІЧНІ ЛЕЙКОЗИ
Парапротеїнемічні лейкози характеризуються здатністю пухлинних клітин синтезувати однорідні імуноглобуліни або їх фрагменти – парапротеїни. При цьому пухлинні клітини є атиповими плазмоцитами і тому зберігають здатність у збоченій формі синтезувати атипові імуноглобуліни.
Мієломна хвороба (плазмоцитома)- хронічний лейкоз, що найчастіше зустрічається серед парапротеїнемічних гемобластозів.
Виникає в основному у дорослих і при сучасних методахлікування може тривати 4-5 років. В основі хвороби лежить пухлинне розростання в кістковому мозку атипових плазмоцитів, які отримали назву мієломних клітин.Вони синтезують парапротеїни, які виявляються в крові та сечі хворих. За характером та поширеністю пухлинного інфільтрату в кістковому мозку виділяють вузлувату та дифузну форми хвороби.
При вузлуватій формі плазмоцитома утворює вузли пухлини у кістковому мозку, зазвичай плоских кісток (зводу черепа, ребер, тазу) та хребців. Лейкозна інфільтрація супроводжується розрідженням кістки або її пазушним розсмоктуванням (остеолізис та остеопороз) з утворенням правильної форми округлих дефектів, які на рентгенограмі виглядають як гладкостінні пробоїни. Пазушне розсмоктування обумовлює вихід кальцію з кісток та розвиток гіперкальціємії з появою множинних вапняних метастазів у м'язах та паренхіматозних органах. Крім того, виникають патологічні переломикісток.
При генералізованій формі мієломної хвороби розростання мієломних клітин відбувається, крім кісткового мозку, у селезінці, лімфатичних вузлах, печінці, нирках та інших внутрішніх органах.
Морфогенез.
У периферичній крові виявляються аномальні імунні білки (парапротеїни), у тому числі дрібнодисперсний білок Бенс-Джонса, який легко проходить через нирковий фільтр та виявляється у сечі. У зв'язку з великою концентрацією білка Бенс-Джонса розвивається парапротеїнемічний нефроз. Крім того, через порушення нормального синтезу імунопротеїнів плазмоцитома часто ускладнюється розвитком амілоїдозу з ураженням нирок. Тому причиною смерті цих хворих нерідко є уремія. Через різке пригнічення функції імунної системи до основного захворювання може приєднуватися вторинна інфекція, яка також спричиняє смерть хворих на мієломну хворобу.
ЗЛОЯКІСНІ ЛІМФОМИ (ГЕМАТОСАРКОМИ)
Злоякісні лімфоми (гематосаркоми)- регіонарні злоякісні пухлини лімфоїдної тканини, що мають моноклонове походження.
Лімфоми розвиваються з незрілих форм лімфоцитів і вражають лімфатичну тканину будь-якої однієї області, однак у термінальній стадії захворювань можлива генералізація пухлинного процесу з розвитком метастазів у кістковий мозок.
Етіологія.
Причини виникнення злоякісних лімфом у принципі не відрізняються від причин, викликають пухлиниіншого походження. Водночас доведено, що частина є лімфом. так само як і деякі інші лейкози, має вірусне походження. Не виключена і спадкова схильність до захворювання. Трансформація нормальних гемопоетичних клітин у пухлинні відбувається в результаті змін у геномі, внаслідок чого нормальна генетична програма гемопоезу змінюється у напрямку пухлинного атипізму.
Класифікація лімфом.
- За клініко-морфологічними особливостями:
- лімфогранулематоз, або хвороба Ходжкіна;
- неходжкінські лімфоми.
- За джерелом зростання (цитогенезу):
- В-лімфоцитарні;
- Т-лімфоцитарні.
- За ступенем диференціювання пухлинних клітин:
- низької злоякісності;
- помірної злоякісності;
- високої злоякісності.
Лімфогранулематоз (хвороба Ходжкіна) описаний 1832 р. англійським лікарем Т. Ходжкіним. Частота захворювання становить 3 випадки на 100 000 населення, або 1% всіх злоякісних новоутворень. Пухлина вражає лімфатичні вузли зазвичай однієї області - шийні, медіастинальні, заочеревинні, рідше пахвові або пахвинні.
Морфогенез.
Уражені лімфатичні вузли збільшуються у розмірах, зливаються між собою та утворюють великі пакети. На початку захворювання лімфатичні вузли м'які, на розрізі рожевого кольору. У міру прогресування лімфоми в них розвиваються некротичні, а потім склеротичні зміни, у зв'язку з чим лімфатичні вузли ущільнюються, на розрізі виглядають сухими та строкатими. У своєму розвитку лімфогранулематоз проходить кілька стадій – від ізольованого ураження групи лімфатичних вузлів до генералізованого ураження внутрішніх органів із придушенням лімфоїдної тканини та заміщенням її полями склерозу.
При мікроскопічному дослідженні пухлина складається з поліморфних пухлинних клітин лімфоцитарного ряду, серед яких є характерні гігантські клітини з лопатевим ядром і вузьким обідком цитоплазми. клітини Березовського-Штернберга. Ці клітини є діагностичною ознакою лімфогранулематозу. Крім того, характерні клітини Ходжкіна - великі клітини з великим світлим ядром та темним ядерцем.
Нерідко у фіналі захворювання воно набуває генералізованого характеру з ураженням багатьох внутрішніх органів - шлунка, легень, печінки, шкіри. При розтині померлих від лімфогранулематозу особливо демонстративно виглядає селезінка - вона збільшена в розмірах, щільна, на розрізі червона з множинними біло-жовтими осередками некрозу та склерозу, що надає їй схожість з особливим виглядомграніту - порфіром(Порфірова селезінка).
Неходжкінські лімфоми.
Це група злоякісних пухлин з недиференційованих та бластних форм В- та Т-клітин лімфатичної тканини. Діагноз цих захворювань вимагає обов'язкового морфологічного та імуногістохімічного дослідження біоптатів лімфатичних вузлів.
 (1оцінок, у середньому: 5,00із 5)
(1оцінок, у середньому: 5,00із 5)