Зернистий орган і зовнішній вигляд. Дистрофія. "зерниста дистрофія" у книгах
Дистрофія- (Від грец. dys - порушення, trophe - харчування) - якісні зміни хімічного складу, фізико-хімічних властивостей та морфологічного виду клітин і тканин організму, пов'язані з порушенням обміну речовин. До дистрофічних процесів немає відношення зміни у метаболізмі і структурі клітин, що відбивають пристосувальну мінливість організму.
Етіологія.
Порушення обмінних процесів, що призводить до структурних змін тканин, спостерігається при дії багатьох зовнішніх і внутрішніх факторів (біологічно неповноцінне годування, різні умови утримання та експлуатації тварин, механічні, фізичні, хімічні та біологічні впливи, інфекції, інтоксикації, порушення крово- та лімфообігу, ураження залоз внутрішньої секреції та нервової системи, генетична патологія та ін.). Патогенні фактори на органи та тканини діють або безпосередньо, або рефлекторно через нервово-гуморальну систему, що регулює обмінні процеси. Характер дистрофічних процесів залежить від сили, тривалості та частоти впливу того чи іншого хвороботворного подразника на організм, а також реактивного стану організму та виду пошкодженої тканини. По суті, дистрофічні зміни відзначають при всіх хворобах, але в одних випадках вони виникають первинно і визначають характер хвороб, а в інших — являють собою неспецифічний або вторинний патологічний процес, що супроводжує захворювання.
Патогенез
. Сучасні методи дослідження (гістохімічні, електронно-мікроскопічні, авторадіографічні, біохімічні та ін.) показали, що в основі будь-якого дистрофічного процесу лежить порушення ферментативних реакцій (ферментопатія) в обміні (синтезі та розпаді) речовин із пошкодженням (альтерацією) структури та функцій клітинно- тканинних систем організму. При цьому в тканинах накопичуються продукти обміну (змінені як кількісно, так і якісно), порушуються фізіологічна регенерація (відновлення живої матерії насамперед на молекулярному та ультраструктурному рівнях її організації) та функції того чи іншого органу, а також життєдіяльність організму загалом.
Механізм розвитку
та сутність змін при різних дистрофіях неоднакові.
За механізмом процесу дистрофічних змін розрізняють декомпозицію; інфільтрацію; трансформацію та змінений, або збочений, синтез.
Декомпозиція (від латів. decompositio — перебудова) — зміна ультраструктур, макромолекул та комплексних (білково-жировуглеводних та мінеральних) сполук клітинних та тканинних систем. Безпосередні причини такої перебудови - порушення балансу поживних речовин, метаболітів та продуктів обміну, гіпоксія та інтоксикація, зміна температури (лихоманка, застуда), порушення кислотно-лужної рівноваги (ацидоз, рідше алкалоз), окислювально-відновного та електролітного потенціалу клітин і тканин. В результаті змін основних параметрів клітинно-тканинних систем (рН, стану АТФ-системи та ін) складні біологічні сполуки клітинних органел і макромолекул або видозмінюються, або розпадаються на простіші сполуки, які стають доступними для гістохімічного дослідження. Вільні білки гідролізуються за участю ферментів лізосом або піддаються денатурації. При цьому поряд з первинним ушкодженням ультраструктур можуть виникати вторинні процеси (наприклад, утворення складних сполук типу амілоїду, гіаліну тощо).
Патологічна інфільтрація(від лат. infiltratio - просочування) характеризується відкладенням і накопиченням (депонуванням) в клітинах і тканинах продуктів обміну (білків, ліпідів, вуглеводів та ін) і речовин, що приносяться зі струмом крові та лімфи ("хвороби накопичення"),
Трансформація (від лат. transformatio - Перетворення) - процес хімічного перетворення сполук в інші, наприклад жирів і вуглеводів в білки або білків і вуглеводів в жири, підвищений синтез глікогену з глюкози і т. д., з надлишковим накопиченням знову утворених сполук.
Змінений синтезбудь-яких сполук виявляється у посиленому чи зменшеному освіті їх із накопиченням чи збіднінням і втратою тканинах, наприклад глікогену, жиру, кальцію та інших. («хвороби недостатності»). Можливий «збочений» (патологічний) синтез з появою та накопиченням у тканинах сполук, не властивих їм в умовах нормального обміну, наприклад синтез незвичайного білка амілоїду, глікогену в епітелії нирок, кератину в епітелії слізної залози, патологічних пігментів та ін.
Зазначені патогенетичні механізми дистрофій можуть виявлятися одночасно чи послідовно з розвитком процесу.
У морфологічному відношеннідистрофії проявляються насамперед порушенням будови ультраструктур клітин та тканин. У фізіологічних умовах перебудова органел клітини та міжклітинної речовини поєднується з процесами їх відновлення, а при дистрофіях порушується регенерація на молекулярному та ультраструктурному рівнях (молекулярний морфогенез). При багатьох дистрофіях у клітинах та тканинах виявляють включення, зерна, краплі або кристали різної хімічної природи, які у звичайних умовах не зустрічаються або їхня кількість збільшується порівняно з нормою. В інших випадках, навпаки, у клітинах та тканинах зменшується кількість властивих їм сполук до повного зникнення (глікогену, жиру, мінеральних речовин та ін.). В обох випадках клітини і тканини втрачають характерну для них тонку структуру (м'язова тканина — поперечну смугастість, залізисті клітини — полярність, сполучна тканина — фібрилярна будова і т. д.), а у важких випадках спостерігається дискомплексація клітинних елементів (наприклад , порушується балкова будова печінки)
Макроскопічні зміни.
При дистрофії змінюються колір, величина, форма, консистенція та малюнок органів. Зміна зовнішнього вигляду органу послужило підставою назвати цей процес переродженням, або дегенерацією - терміном, що не відображає сутність дистрофічних змін.
Функціональне значення дистрофії.
Полягає воно у порушенні основних функцій органу (наприклад, синтез білка, вуглеводів, ліпопротеїдів при гепатозі, протеїнурія при нефрозі, ослаблення серцевої діяльності при дистрофії міокарда тощо). Після усунення причини, що викликала розвиток дистрофічного процесу, обмін речовин у клітинах, тканинах та цілому організмі, як правило, нормалізується, внаслідок чого орган набуває функціональної повноцінності та звичайного зовнішнього вигляду. Однак важкі дистрофічні зміни бувають незворотними, тобто наростаюча диспропорція між підвищеним розпадом власних структур і недостатнім відновленням закінчується їх некрозом.
БІЛКОВА ДИСТРОФІЯ (диспротеїноз)
Білкові дистрофії- Структурно-функціональні порушення тканин, пов'язані зі змінами хімічного складу, фізико-хімічних властивостей та структурної організації білків. Виникають вони у разі порушення рівноваги між синтезом і розпадом білків у клітинах і тканинах внаслідок білкової чи амінокислотної недостатності, на час вступу до тканини чужорідних для організму речовин, і навіть при патологічному синтезі білків. Порушення білкового обміну в організмі різноманітні. Вони можуть мати місцеве чи загальне (системне) поширення. По локалізації розрізняють порушення білкового обміну в клітинах (клітинні, або паренхіматозні, диспротеїнози), у міжклітинній речовині (позаклітинні, або стромально-судинні, диспротеїнози) або одночасно в клітинах та міжклітинній речовині (змішані диспротеїнози).
КЛІТИННІ (ПАРЕНХІМАТОЗНІ) ДИСПРОТЕЇНОЗИ
Зерниста дистрофія, або каламутне набухання, - Порушення колоїдних властивостей та ультраструктурної організації клітин з виявленням білка у вигляді зерен. Це найчастіший вид білкових дистрофій.
Причини: інфекційні та інвазійні хвороби, неповноцінне годування та інтоксикації, розлади крово- та лімфообігу та інші патогенні фактори.
Патогенезскладний. Провідний механізм - декомпозиція, в основі якої лежить недостатність АТФ-системи, пов'язана з гіпоксією, дією токсичних речовин на ферменти окисного фосфорилювання (ферментопатія). Внаслідок цього знижується окислювально-відновний потенціал клітин, накопичуються недоокислені та кислі (ацидоз), рідше лужні (алкалоз) продукти обміну, збільшуються онкотично-осмотичний тиск та проникність мембран. Розлад електролітного та водного обмінів супроводжується набуханням білків клітин, порушенням ступеня дисперсності колоїдних частинок та стійкості колоїдних систем, особливо в мітохондріях. У цьому зростає активність гідролітичних ферментів лизосом. Гідролази розривають внутрішньомолекулярні зв'язки шляхом приєднання молекул води, викликаючи перебудову комплексних сполук та макромолекул. Адсорбція будь-яких токсичних речовин у ліпопротеїдних та глікопротеїдних комплексах викликає також їх перебудову та розпад. Білок, що звільняється, а потім і інші компоненти комплексних сполук (жир та ін) укрупнюються, а будучи в ізоелектричному стані, коагулюють з появою зерен. При цьому можливо порушується синтез білка цитоплазми (молекулярний морфогенез), як це було показано за допомогою мічених атомів (С. В. Анічков, 1961).
Поряд з декомпозицією поява зернистості пов'язана також з патологічною трансформацією вуглеводів та жирів у білки, інфільтрацією та резорбцією чужорідних для організму білків (парапротеїдів), що приносяться струмом крові (диспротеїнемія).
Гістологічні ознакизернистої дистрофії найбільш яскраво виражені у печінці, нирках, міокарді, а також у скелетних м'язах (тому її називають ще паренхіматозною). Відзначають нерівномірне збільшення обсягу епітеліальних клітин і м'язових волокон, що здавлюють капіляри, набухання і помутніння цитоплазми, згладженість і зникнення тонкої структури (щіткової облямівки залозистого епітелію, поперечної смугастість у м'язовій тканині і т. д.), поява і природи. У цьому межі клітин та обриси ядер помітні важко. Іноді цитоплазма набуває пінистого вигляду, деякі клітини відокремлюються від базальної мембрани та одна від одної (дискомплексація). Під впливом слабкого розчину оцтової кислоти чи лугу цитоплазма просвітлюється, ядро стає знову помітним. Поряд із розчинністю у слабких кислотах та лугах наявність білка у зернах визначають гісто-хімічними методами, а також за допомогою електронного мікроскопа.
Електронно-мікроскопічнозерниста дистрофія характеризується набуханням та округленням мітохондрій, розширенням цистерн та трубочок цитоплазматичної мережі. Мітохондрії збільшуються, мембрани їх розтягуються, розшаровуються, гребінці нерівномірно товщають і коротшають, структурні білки мітохондрій розчиняються з просвітленням матриксу і появою прозорих вакуолей (вакуолізація мітохондрій) або набухають і укрупняються. Розпадається також білково-синтезуючий апарат клітини (полісоми, рибосоми).
Макроскопічноуражені органи збільшені в обсязі, в'ялої консистенції, малокровні, на розрізі тканина вибухає за межі капсули, поверхня розрізу тьмяна, печінка і нирки сірувато-коричневого кольору зі згладженим малюнком, а м'язова тканина (міокард, скелетні м'язи) нагадує ошпарене.
Клінічне значеннязернистої дистрофії у тому, що порушуються і можуть змінюватися якісно функції уражених органів (серцева слабкість при інфекційних хворобах, альбумінурія при поразці нирок тощо. буд.).
Вихідзалежить від багатьох причин. Зерниста дистрофія відноситься до оборотних процесів, але якщо її причини не усунуті, то на висоті розвитку вона може переходити в більш важкий патологічний процес - в гідропічну, гіаліново-краплинну, жирову та інші види дистрофій з наслідком некрозу клітини (так звана ацидофільна дегенерація , «Балонна» дистрофія або коагуляційний некроз).
Диференційна діагностика. Зернисту дистрофію необхідно відрізняти від фізіологічного синтезу білків у клітині з накопиченням білкової зернистості, пов'язаної з нормальною життєдіяльністю організму (наприклад, утворення гранул секрету в залозистому органі) або фізіологічною резорбцією клітинного білка (наприклад, у ниркових канальцях проксимального сегмента). Від посмертної зміни органів (трупної тьмяності) цей прижиттєвий процес відрізняється ясно вираженим збільшенням розмірів клітини та органів, а також нерівномірністю патологічних уражень.
Гіаліново-крапельна дистрофія(від грец. hyalos - склоподібний, прозорий) - внутрішньоклітинний диспротеїноз, що характеризується появою в цитоплазмі прозорих оксифільних білкових крапель.
Причини:гострі та хронічні інфекції, інтоксикації та отруєння (сулемою, солями хрому, урану тощо); крім того, дистрофія може бути наслідком алергічних процесів після попередньої сенсибілізації білками. Її відзначають також при хронічних катарах шлунково-кишкового тракту, сечового міхура, актиномікомах і пухлинах.
Патогенез- Гіаліново-крапельної дистрофії полягає в тому, що в патологічних умовах відбувається глибока денатурація ліпопротеїдів цитоплазми з випаданням грубої дисперсної фази внаслідок втрати білком гідрофільних властивостей. В інших випадках можливі резорбція та патологічна інфільтрація клітини грубодисперсними чужорідними для організму білками – парапротеїдами, що надходять із крові.
Макроскопічногіаліново-краплинну дистрофію не діагностують.
Гістологічні змінизустрічаються в залізистих органах (печінка та ін), пухлинах, м'язовій тканині, а також в осередках хронічного запалення, але особливо часто-в епітелії канальців нирок. При цьому в цитоплазмі видно більш менш однорідні, напівпрозорі краплі білка, що фарбуються кислими барвниками (наприклад, еозином). У міру накопичення крапель та злиття їх між собою вони можуть повністю заповнювати клітину. Найбільш важкі зміни бувають при гломерулонефритах та білковому нефрозі в епітелії звивистих канальців. Подібні зміни виникають в епітелії надниркових залоз і бронхів. У хронічно запалених тканинах, переважно у плазмоцитах, знаходять так звані русселівські, або фуксинофільні, тільця у вигляді великих гомогенних, іноді шаруватих гіалінових куль, які інтенсивно фарбуються фуксином і після розпаду клітин лежать вільно у тканині. Електронно-мікроскопічно відзначають появу гіалінових крапель і вакуолей у цитоплазмі, набухання та розпад мітохондрій, зникнення полісом та рибосом, розрив цистерн мережі та ін.
Клінічне значеннягіаліново-крапельної дистрофії в тому, що вона відображає різко виражену недостатність органу, зокрема нирок.
Вихід.У зв'язку з незворотною денатурацією плазмового білка гіаліново-краплинна дистрофія протікає з наслідком некрозу.
Гідропічна (водяна, вакуольна) дистрофія- Порушення білково-водноелектролітного обміну клітини з вивільненням всередині клітин води.
Причини:інфекційні хвороби (ящур, віспа, вірусний гепатит та ін), запальна інфільтрація тканин, фізичні, хімічні та гострі токсичні впливи, що викликають гіпоксію та розвиток набряку, хвороби обміну речовин (білкова недостатність, сольове голодування, гіповітамінози, наприклад пелагра, та ін. ), а також хронічні інтоксикації та виснаження (хронічні гастроентерити, коліт та ін.).
Патогенез.В результаті зниження окисних процесів, нестачі енергії та накопичення недоокислених продуктів обміну зв'язана вода не тільки звільняється і затримується в клітині (інтрацелюлярна вода), але і надходить з тканинної рідини в клітину (екстрацелюлярна вода) у зв'язку з підвищенням колоїдно-осмотичного тиску та порушенням проникності клітинних мембран. При цьому іони калію виходять із клітини, тоді як іони натрію посилено проникають у неї внаслідок порушення процесів осмосу, пов'язаних із «іонним насосом». Біохімічна сутність дистрофій полягає в активізації гідролітичних ферментів лізосом (естераз, глюкозидаз, пептидаз та ін.), що розривають внутрішньомолекулярні зв'язки шляхом приєднання води, викликаючи гідроліз білків та інших сполук.
Гістологічні зміничасто встановлюють в епітеліальній тканині шкірного покриву, печінки, нирок, надниркових залоз, нервових клітинах, м'язових волокнах і лейкоцитах. Вони спостерігають ознаки зернистої дистрофії, часткового цитолиза із заснуванням у цитоплазмі вакуолей (вакуольна дистрофія), наповнених рідиною, що містить білок і ферменти. Іноді білок цитоплазматичної рідини згортається під впливом солей кальцію. Подальше розчинення цитоплазми та збільшення кількості води у ній викликають більш виражений внутрішньоклітинний набряк, розвиток якого може призвести до каріоцитолізу. Клітина при цьому збільшується, ядро та цитоплазма розчиняються, зберігається лише її оболонка. Клітина набуває вигляду балона (балонна дистрофія). Електронно-мікроскопічно відзначають розширення та розрив цистерн та трубочок, набухання та лізис мітохондрій, рибосом та інших органел, а також розчинення основної плазми.
Макроскопічнооргани та тканини змінюються мало, за винятком набряклості та блідості їх. Вакуольну дистрофію визначають лише під мікроскопом.
Клінічне значеннягідропічної дистрофії у цьому, що знижуються функції ураженого органа.
Вихід. Вакуольна дистрофія є оборотною за умови, якщо немає повного розчинення цитоплазми клітини. При збереженні ядра та частини цитоплазми нормалізація водно-білкового та електролітного обмінів призводить до відновлення клітини. При значному руйнуванні органел з розвитком вираженого набряку (балонної дистрофії) настають незворотні зміни (колікваційний некроз).
Вакуольну дистрофію необхідно відрізняти від жирової, використовуючи гістохімічні методи визначення жиру, так як у процесі виготовлення гістопрепаратів із застосуванням розчинників (спирту, ефіру, ксилолу, хлороформу) жирові речовини витягуються і їх місці також з'являються вакуолі.
Рогова дистрофія чи патологічне орговіння- Надлишкове (гіперкератоз) або якісно порушене (паракератоз, гіпокератоз) утворення рогової речовини. Кератин забарвлюється еозином у рожевий колір, а пікрофуксином за Ван Гізоном – у жовтий. Він має осміофільність і високу електронну щільність.
Причини:порушення обміну речовин в організмі - білкова, мінеральна (недолік цинку, кальцію, фосфору) або вітамінна недостатність (гіповітаміноз А, особливо у птахів, великої рогатої худоби та свиней, пелагра та ін.); інфекційні хвороби, пов'язані із запаленням шкіри (дерматофітози, короста, парша та ін.); фізичні та хімічні дратівливі впливи на слизові оболонки та шкіру; хронічне запалення слизових оболонок; іноді спадкові захворювання (іхтіоз - утворення рогових нашарувань на шкірі, що нагадують риб'ячу луску або панцир черепахи). Надмірне утворення рогу спостерігають у бородавках, канкроїді (ракоподібної пухлини) та дермоїдних кістах.
Патогенезрогової дистрофії пов'язаний з надлишковим або порушеним синтезом керотину в епідермісі шкіри і в епітелії, що орогів, слизових оболонок. Утворення рогової речовини в слизових оболонках травного тракту, верхніх дихальних шляхів і статевих органів супроводжується заміною залозистого епітелію ороговіючим плоским багатошаровим.
Паракератоз(від грецьк. para - навколо, keratos - рогова речовина) виявляється у втраті здатності клітин епідермісу виробляти кератогіалін.
Гістологічнопри паракератозі виявляють потовщення епідермісу внаслідок гіперплазії клітин мальпігієвого шару та надмірного накопичення рогової речовини. У слизових оболонках шкірного типу та в епідермісі шкіри можливе сосочкове потовщення епідермісу через гіперплазію шару шилоподібних клітин та подовження шилоподібних відростків. Такі поразки називають акантозом (від грец. Akantha - Шип, голка).
При пара-і гіпокератозі виражена атрофія зернистого шару, роговий шар пухкий, з дискомплексованими клітинами, що мають паличкоподібні ядра (неповне зроговіння).
Макроскопічноу місцях патологічного зроговіння (поширеного чи місцевого) шкіра потовщена, з надмірним розростанням рогового шару. Вона втрачає еластичність, стає шорсткою і жорсткою, утворюються сухі потовщення та мозолі. При паракератозі роговий шар потовщений, пухкий, з підвищеним лущенням рогових лусочок, іноді випаданням волосся. У дорослих тварин, особливо у молочних корів, відзначають неправильне зростання копитного рогу, яке втрачає глазур і розтріскується.
При лейкоплакії (від грец. leukos - білий, plax, axos - плита) на слизових оболонках утворюються різного розміру вогнища ороговілого епітелію у вигляді тяжів і бляшок сіро-білуватого кольору.
Клінічне значенняПатологічного зроговіння пов'язано з розвитком інфекційних ускладнень. Лейкоплакія може стати джерелом розвитку епітеліальних пухлин (папілом, рідше раку).
Вихідрогової дистрофії залежить від перебігу основної хвороби. При усуненні причини, що викликає патологічне зроговіння, пошкоджена тканина може відновлюватися. Новонароджені тварини, які страждають на їхтіоз, зазвичай гинуть у перший день життя.
Позаклітинні (струмально-судинні) диспротеїнози
Це порушення білкового обміну в міжклітинній речовині. Сутність їх полягає в патологічному синтезі білків клітинами мезенхімального походження, в дезорганізації (розпаді) основної речовини та волокнистих структур з підвищенням судинно-тканинної проникності та накопиченням у міжклітинній речовині сполучної тканини білків крові та лімфи, а також продуктів метаболізму. Ці процеси можуть бути місцевими чи поширеними. До них відносяться борошно набухання, фібриноїдне набухання (фібриноїд), гіаліноз і амілоїдоз.
Мукоїдне набухання- Початкова стадія дезорганізації сполучної тканини (строми органів, судин), яка характеризується порушенням зв'язку з протеїнами та перерозподілом кислих глікозаміногліканів (гіалуронової, хондроїтинсерної кислот та ін.).
Причини:кисневе голодування, інтоксикації, деякі хвороби обміну речовин (гіповітамінози С, Е, К) та ендокринної системи (мікседема), алергічні гострі та хронічні хвороби сполучної тканини та судин («колагенові хвороби», ревматизм, атеросклероз та ін.), в етіологічну роль грає гемолітичний стрептокок групи А, а також інфекційні хвороби (набрякова хвороба поросят, бешиха свиней та ін).
Патогенеззмін при мукоидном набуханні полягає у порушенні синтезу міжклітинної речовини або в її поверхневому розпаді під дією гіалуронідази екзогенного (гемолітичний стрептокок та ін) або ендогенного походження, а також в умовах наростаючої гіпоксії тканини з розвитком ацидозу середовища. Це веде до деполімеризації білково-полісахаридного комплексу та накопичення вивільнених кислих глікозоаміногліканів (особливо гіалуронової та хондроїтинсерної кислот), які, володіючи гідрофільними властивостями, викликають підвищення тканинної та судинної проникності, серозний набряк тканини (серозний набряк). опротеїдів).
Мікроскопічномукоїдне набухання сполучної тканини визначається базофілією і метахромазією волокон і основної речовини (наприклад, толуїдиновий синій забарвлює кислі глікозаміноглікани в червоний колір, пікрофуксин - не в червоний, а в жовто-оранжевий колір). Сутність метахромазії (від грец. metha - Зміна, chromasia - фарбування) полягає в здатності глікозоаміногліканів викликати полімеризацію барвника. І якщо барвник як мономер має синій колір, як димер, тріммер - фіолетовий, то як полімер - червоний (таутомерія). Зміни молекулярної структури колагенових волокон супроводжуються їх набуханням, нерівномірно вираженим збільшенням обсягу та розмитістю контурів та структури, розволокненням, а зміна проміжної речовини – скупченням Т-лімфоцитів та гістіоцитів.
Макроскопічноорган залишається без зміни, але опорно-трофічні та бар'єрні функції сполучної тканини порушуються.
Вихід. Можливе повне відновлення пошкоджених структур або перехід у фібриноїдне набухання.
Фібриноїдне набухання- Глибока дезорганізація сполучної тканини строми органів, судин, що характеризується посиленою деполімеризацією білків-полісахаридних комплексів основної речовини та фібрилярних структур з різким підвищенням судинно-тканинної проникності. У зв'язку з плазморагією сполучна тканина просочується білками крові (альбумінами, глобулінами, глікопротеїдами, фібриногеном). В результаті преципітації або хімічної взаємодії цих сполук утворюється складна в хімічному відношенні неоднорідна речовина - фібриноїд, до складу якого входять білки і полісахариди колагенових волокон, що розпадаються, основної субстанції і плазми крові, а також клітинні нуклеопротеїди.
Причини:ті ж алергічні, інфекційні фактори, нейротрофічні порушення, які викликають мукоїдне набухання, але діють з більшою силою або тривалістю. Як місцевий процес фібриноїдне набухання спостерігається в осередках хронічного запалення.
Патогенез.Фібриноїдні зміни, будучи наступною стадією мукоидного набухання, розвиваються в тому випадку, якщо процес дезорганізації сполучної тканини поглиблюється, відбуваються розпад не тільки основної речовини, але також колагенових та інших фібрилярних структур, деполімеризація глікозоаміногліканів, колагенових волокон, що розпадаються, числа грубодисперсним білком - фібриногеном, що є обов'язковим компонентом фібриноїду. При цьому порушується фібриллогенез, особливо біосинтез кислих глікозаміногліканів у мезенхімних клітинах, а також спостерігається проліферація Т-лімфоцитів та гістіоцитів. Хімічна взаємодія та полімеризація продуктів розпаду основної речовини, колагену та білків плазми супроводжуються утворенням незвичайних білково-полісахаридних комплексів фібриноїду.
Гістологічні змінипротікають у дві стадії: фібриноїдне набухання та фібриноїдний некроз. При фібриноїдному набуханні відзначають розпад основної речовини, набухання та частковий розпад колагенових та еластичних волокон, плазморагію з просочуванням сполучної тканини альбумінами, глобулінами плазми та фібриногеном, який виявляється гістохімічними та імунофлуоресцентними. Колаген, утворюючи з фібриногеном та іншими речовинами щільні нерозчинні сполуки, змінює свої тинкторіальні властивості: він стає еозино-, піроніно- та аргірофільним, пікрофуксином фарбується у жовтий колір, ШІК-реакція різко позитивна. Процес завершується повною деструкцією сполучної тканини з розвитком фібриноїдного некрозу. При цьому тканина набуває вигляду зернисто-глибинної або аморфної маси, до складу якої входять продукти розпаду колагенових волокон, основної речовини та плазмових білків. При повній деполімеризації вільних глікозаміногліканів метахромазія зазвичай не виражена. Навколо некротичних мас розвивається продуктивне запалення з утворенням неспецифічних гранульом, що складаються з Т-лімфоцитів та макрофагів.
Макроскопічнофібриноїдні зміни сполучної тканини малопомітні, їх виявляють під мікроскопом.
Клінічне значенняфібриноїдного набухання випливає із порушення або виключення функції ураженого органу.
Вихідпов'язаний із перебігом основної хвороби, при якій розвивається цей процес. Фібриноїдні маси можуть резорбуватися, заміщатися сполучною тканиною, яка піддається склерозу або гіалінозу.
Гіаліноз(від грец. hyalos - прозорий, склоподібний), або гіалінова дистрофіясвоєрідне фізико-хімічне перетворення сполучної тканини у зв'язку з утворенням складного білка - гіаліну, подібного за морфологічними ознаками з основною речовиною хряща. Гіалін надає тканинам особливого фізичного стану: вони стають гомогенними, напівпрозорими і щільнішими. До складу гіаліну входять глікозоаміноглікани та білки сполучної тканини, плазми крові (альбуміни, глобуліни, фібриноген), а також ліпіди, солі кальцію. Дані електронної мікроскопії свідчать, що у складі гіаліну є різновид фібрилярного білка (фібрину). Гіалін стійкий до дії кислот, лугів, ферментів, що інтенсивно забарвлюється кислими барвниками (еозином, кислим фуксином або пікрофуксином) в червоний або жовтий колір, дає ШІК-позитивну реакцію.
Причини.Гіаліноз розвивається у результаті різних патологічних процесів: плазматичного просочування, мукоидного і фібриноїдного набухання сполучної тканини. Фізіологічний прототип гіалінозу – старіння.
Системний гіалінозсудин та сполучної тканини спостерігається при колагенових хворобах, артеріосклерозі, інфекційних та токсичних хворобах, хронічному запаленні, хворобах, пов'язаних з порушенням білкового обміну, особливо у високопродуктивних корів та свиней. Виражений гіаліноз судин трапляється при хронічному гломерулонефриті, особливо у собак. Поряд з цим місцевий гіаліноз (склероз) зустрічається у новоствореній сполучній (рубцевій) тканині.
Патогенез.У виникненні та розвитку системного гіалінозу важливу роль відіграють гіпоксія тканин, пошкодження ендотелію та базального шару судинної стінки, порушення синтезу та структури ретикулярних, колагенових, еластичних волокон та основної речовини сполучної тканини. При цьому відбуваються підвищення судинної та тканинної проникності, просочування тканини плазмовими білками, їх адсорбція з утворенням складних білкових сполук, преципітація та ущільнення білкових мас.
У розвитку гіалінозу беруть участь також імунологічні механізми, оскільки доведено, що гіалінові маси мають деякі властивості імунних комплексів антиген-антитіло.
Гістологічногіалін виявляють у міжклітинній речовині сполучної тканини. Системний гіаліноз стінок кровоносних судин та сполучної тканини проявляється утворенням гіаліну в основному речовині інтими та периваскулярної сполучної тканини артерій та капілярів. Зрештою утворюється гомогенна щільна білкова маса, що фарбується кислими барвниками. Хоча гіалін є індиферентною речовиною, але накопичення його супроводжується потовщенням стінки судини, витісненням медії гіаліновою масою зі звуженням просвіту, аж до повного закриття (облітерації) його в дрібних судинах. Некротизація тканин, що піддаються гіалінозу, може супроводжуватися їх кальцинацією, розривами стінки судини з виникненням крововиливів і тромбозів. У залізистих органах гіаліноз сполучної тканини супроводжується потовщенням базальних мембран залоз, стисканням залізистого епітелію з подальшою його атрофією. Місцевий гіаліноз зустрічається в осередках хронічного запалення, у новоствореній сполучній тканині (сполучнотканинних капсулах і старих рубцях). При цьому колагенові волокна набухають, зливаються в однорідні тканини, а атрофуються клітини.
Макроскопічнооргани і тканини, уражені гіалінозом у слабкій мірі, не мають помітно виражених змін, процес виявляють лише під мікроскопом. При різко вираженому гіалінозі судини втрачають еластичність, а уражені органи стають блідими та щільними. При випадінні солей кальцію в гіалінові маси вони ще більше ущільнюються.
Функціональне значеннягіалінозу залежить від ступеня та поширеності його. Системний гіаліноз спричиняє порушення функції органів, особливо їх судин, з розвитком атрофії, розриви та інші тяжкі наслідки. Місцевий гіаліноз може викликати істотних функціональних змін.
Вихідрізний. Встановлено, що гіалінові маси можуть розпушуватися і розсмоктуватися або ослизняться, наприклад, у рубцях, так званих келоїдах. Однак у більшості випадків поширений гіаліноз проявляється як незворотний процес.
Диференційна діагностика.Патологічний гіаліноз слід відрізняти від фізіологічного, який проявляється у процесі інволюції та нормального старіння тканин (наприклад, інволюція жовтого тіла, судин матки, молочної залози тощо). При цьому гіаліноз матки та молочної залози звернений у зв'язку з посиленням функції органу. Зовні з гіалінозом подібне гіаліно-подібне перетворення мертвих тканин, продуктів секреції (наприклад, утворення гіалінових циліндрів при нефроз-нефриті, гіалінових тромбів, гіалінізація фібрину і т. д.).
Амілоїдоз (амлоїдна дистрофія)характеризується патологічним синтезом своєрідного фібрилярного білка (преамілоїду) у клітинах ретикулоендотеліальної системи з подальшим утворенням амілоїду – складного глікопротеїду. Р. Вірхов (1859) прийняв цей глікопротеїд за крохмалоподібну сполуку (amylum — крохмаль) через характерне фарбування його в синій колір йодом та сірчаною кислотою. У зв'язку з міцністю хімічних зв'язків амілоїд стійкий до дії кислот, лугів, ферментів, що протистоїть гниття. Кислі глікозоаміноглікани (хондроїтинсульфат) з різним ступенем полімеризації надають амілоїду властивість метахромазії, що відрізняє його від гіаліну та інших білків. Амілоїд забарвлюється генціан- і крезілвіолет в рожево-червоний колір на фіолетовому тлі тканини. Йодгрюн також забарвлює амілоїд у червоний, а конго червоний – у буро-коричневий колір. Конго червоний, введений у кров, здатний накопичуватися в амілоїдній масі in vivo, що використовується для діагностики прижиттєвої амілоїдозу. Амілоїдні маси дають ШІК-позитивну реакцію. Хімічний склад амілоїду може бути різним. У зв'язку з цим деякі барвисті реакції амілоїду (наприклад, метахромазія) випадають (парамілоїд).
Причини системного амілоїдозу:запальні, нагноєльні, некротичні процеси будь-якого походження та інтоксикації. У цих випадках амілоїдоз розвивається як ускладнення хвороби (вторинний або типовий амілоїдоз), спричинене розпадом тканинного білка (наприклад, при туберкульозі, злоякісних пухлинах, неспецифічних запальних процесах з нагноєнням та ін.). Вторинний амілоїдоз спостерігається у лактуючих високопродуктивних корів, птахів, хутрових звірів, коней («сінна хвороба») та ін. Причини атипового первинного (ідіопатичного) та старечого амілоїдозу, характерного для людини, невідомі. Генетичний амілоїдоз є спадковою ензимопатією або аномалією (мутацією) в генетичному апараті клітин РЕМ. В експерименті на лабораторних тваринах амілоїдоз можна викликати парентеральним введенням чужорідного білка (казеїну), а також шляхом створення вогнищ хронічного нагноєння. У зв'язку з тривалим парентеральним запровадженням чужорідного білка розвивається амілоїдоз у коней - продуцентів імунних сироваток.
Причини місцевого амілоїдозу: хронічні запальні процеси із застоєм крові та лімфи.
Патогенезамілоїдозу складний.
За теорією диспротеїнозу(К. Apitz, Е. Randerath, 1947) Амілоїд виникає на основі порушеного білкового синтезу з появою в крові парапротеїнів або параглобулінів і розвитком диспротеїнемії та гіпергамма-глобулінемії. Ці продукти грубодисперсної білкової фракції плазми крові, виділяючись через ендотеліальний бар'єр, насамперед у селезінці, печінці та нирках, вступають у поєднання з кислими глікозаміногліканами, які звільняються під впливом плазмових білків і тканинних гіалуронідаз, і утворюють.
За теорією аутоімунітету(Loeschke, Letterer, 1962) вирішальне значення в утворенні амілоїду мають змінена реактивність організму та аутоімунні процеси. При багатьох процесах, що ускладнюються амілоїдозом, накопичуються продукти розпаду тканин, лейкоцитів, бактерій, які мають антигенні властивості. Можливо, що порушення реакцій в імунній системі, пов'язані з надлишком антигену та нестачею антитіл, призводять до появи в крові специфічних для тканинних білків преципітинів та фіксації білкового комплексу в місцях утворення антитіл (Letterer). Ця теорія зберегла своє значення для експериментального та вторинного амілоїдозу. Механізм розвитку ідіопатичного, генетичного і старечого амілоїдозу вона не пояснює.
Теорія клітинного локального генезу(G.Teilum, 1962) розглядає амілоїд як продукт білкового синтезу клітинами мезенхімальної системи з перекрученим метаболізмом («мезенхімальна хвороба»). Вона підтверджується вибірковістю ураження цієї системи та внутрішньоклітинним утворенням фібрил преамілоїду клітинами мезенхімальної природи.
Висувається нова мутаційна теорія амілоїдозу(Е. Benditt, N. Eriksen, 1977; В. В. Сєров, І. А. Шамов, 1977), яка може стати універсальною для розуміння патогенезу всіх відомих його форм з
враховуючи різноманітність факторів, що викликають мутацію. За цією теорією клітини, що мутуються, не розпізнаються імунокомпетентною системою і не елімінуються, тому що амілоїдні фібрили є надзвичайно слабкими антигенами. Реакція резорбції амілоїду (амілоїдоклазія), що з'являється, на самому початку його утворення буває недостатньою і швидко пригнічується. Виникають імунологічна толерантність (терпимість) організму до амілоїду та незворотний розвиток амілоїдозу. Мутаційна теорія пояснює близькість амілоїдозу до пухлинних процесів.
Гістологічні та макроскопічні змінизалежать від причини утворення, відношення до різних сполучнотканинних клітин і локалізації амілоїду.
При загальному типовому амілоїдозі, найбільш поширеному у сільськогосподарських тварин, амілоїд випадає по ходу ретикулярних волокон судинних та залозистих мембран та в периретикулярні простори паренхіматозних органів (периретикулярний або паренхіматозний амілоїдоз). Уражаються печінка, селезінка, нирки, рідше надниркові залози, гіпофіз, власна оболонка залоз кишечника, інтима капілярів та артеріол. У сполучнотканинних клітинах накопичуються преамілоїдні фібрили, зникають рибосоми, гіпертрофуються мітохондрії (гігантські мітохондрії), а також пластинчастий комплекс Гольджі (А. Полікар, М. Бессі, 1970).
Накопичення амілоїду в тканині супроводжується атрофією та загибеллю паренхіматозних елементів органу.
Амілоїдоз печінкихарактеризується утворенням амілоїду в навколо синусоїдному просторі (просторі Дисе) між зірчастими ретикулоендотеліоцитами та печінковими клітинами (рис. 8). Амілоїд відзначають також у стінках міждолькових капілярів та артеріол. У міру накопичення амілоїдної речовини печінка збільшується в розмірі, набуває блідо-коричневого кольору, більш щільну, а у коней в'ялу консистенцію. У коней вона може досягати маси 16-33 кг, при цьому близько 10% випадків закінчуються розривом печінки у зв'язку з розплавленням строми (А. П. Гіндін, 1959), з'являються синці, які нерідко закінчуються смертельним крововиливом в черевну порожнину.
Амілоїдоз селезінкипроявляється у двох формах: фолікулярної та дифузної. У першому випадку амілоїд відкладається в ретикулярну тканину фолікулів, починаючи з їхньої периферії. Ретикулярна та лімфоїдна тканини фолікулів атрофуються та заміщуються амілоїдними масами. Макроскопічно амілоїдно змінені фолікули на розрізі мають вигляд напівпрозорих зерен, що нагадують зерна розвареного саго («сагова селезінка»). У другому випадку амілоїд випадає більш менш рівномірно по всій ретикулярній стромі органу і під ендотелією синусів. При дифузному амілоїдозі селезінка збільшена у розмірі, щільної консистенції, а у коней тестуватою; поверхня розрізу гладка, світло-червоно-коричнева, нагадує собою сиру шинку («сальна», або «шинкова», селезінка). У коней можливі розрив органу та крововиливу.
У ниркахамілоїд відкладається в першу чергу в мезангіумі та за ендотелієм капілярних петель та артеріол клубочків, а також у ретикулярній стромі кіркової та мозкової речовин, у стінках артеріол та дрібних артерій, рідше в базальному шарі під епітелієм канальців. Ниркові клубочки поступово атрофуються, епітелій канальців, крім того, піддається зернистій та гіаліново-крапельній дистрофії. У міру накопичення амілоїду нирки збільшуються у розмірі, стають блідо-коричневими, воскоподібними, сухими. При ізольованому ураженні ниркових клубочків вони мають вигляд сірувато-червоних цяточок.
В інших органах(Надниркові залози, гіпофіз, кишечник) амілоїд відкладається в ретикулярній стромі і базальному шарі судин і залоз. У зв'язку з тим, що органи при амілоїдозі набувають воскового або сального вигляду, угорський патолог К. Рокитанський в 1844 р. описав ці зміни під назвою сальної хвороби.
Первинний атиповий амілоїдозз системним ураженням адвентиції судин середнього та великого калібрів, міокарда, попереч-носмугастих і гладких м'язів, шлунково-кишкового тракту, легенів, нервів, шкіри у сільськогосподарських тварин - порівняно рідко"» явище. Його відзначають при хворобах сполучної тканини інфекційно-алергічного При цьому амілоїд виявляється головним чином у стінках капілярів і артерій, у плазмалем фібробластів і колагенових волокон (периколлагеновий амілоїдоз). схильність до розвитку клітинно-проліферативної реакції з утворенням вузлуватих розрощень.
До рідкісних атипових форм амілоїдозу відноситься місцевий амілоїдозз відкладенням амілоїдних мас у сполучну тканину та у стінку судин на ізольованій ділянці органу. Його зустрічають в альвеолах легень при хронічній пневмонії, у слизовій оболонці носової порожнини у коней, у передміхуровій залозі у старих тварин (собаки та ін.), у центральній нервовій системі на місці дистрофічно змінених та загиблих нервових клітин, а також у слизових оболонках інших органів .
Функціональне значенняамілоїдозу пов'язано з розвитком атрофії та загибеллю паренхіматозних клітин та прогресуючої недостатності органу (печінкова, ниркова), розладом крово- та лімфообігу та можливістю розриву органу (зокрема, у коней), що супроводжується іноді смертельною кровотечею.
Вихідзагального амілоїдозу зазвичай буває несприятливим. Однак є експериментальні, клінічні та патоморфо-логічні дані про те, що амілоїдні маси можуть розсмоктуватися за участю гігантських клітин, якщо причина його утворення усунена (М. Н. Нікіфоров, А. І. Струков, Б. І. Мигунов, 1971). У тварин амілоїдоз належить до незворотних процесів.
Гематиниє окислювальною формою гема. Вони мають вигляд анізотропних зерен або кристалів темно-коричневого кольору, містять залізо у зв'язаному вигляді, знебарвлюються перекисом водню, розчиняються у лугах, у кислотах малорозчинні. До них відносять пігменти: малярійний (гемомеланін), соляно-кислий (гемін) та формаліновий. У зв'язку з утворенням великої кількості кров'яних пігментів селезінка, кістковий мозок і печінка можуть набувати аспідно-сіре забарвлення. Солянокислий гематин утворюється при дії на гемоглобін ферментів шлункового соку та соляної кислоти, надаючи ерозіям та виразкам шлунка, а також його вмісту за наявності крововиливів буро-чорний колір. Формаліновий пігмент зустрічається в багатих кров'ю тканинах при фіксації їх у кислому формаліні. Він випадає у вигляді зерен, глибок чи тонких темно-коричневих кристалів. Пігмент пропадає після обробки гістозрізу слабким (1-2%-ним) водним або спиртовим (50-70%-ним) розчином їдкого лугу (КОН).
Порфірини- Попередники гема, позбавлені заліза. Надмірне накопичення їх у крові (порфіринемія) супроводжується розвитком гемолітичної анемії та спленомегалії, коричнево-жовтої або майже чорної пігментації нирок (порфіринурія з наявністю червоної сечі), кістяка (остеогемохроматоз) та дентину зубів у свиней та великого. Зерна пігменту виділяються в клітинах мононуклеарно-макрофагальної системи кісткового мозку та в епітелії сечових канальців нирок, надаючи їм радіальну смугастість. Розвиток вродженої (ідіопатичної) порфірії пов'язане з блокуванням в еритроцитах ферментативного перетворення протопорфірину III в уропорфірину III, що лежить в основі будови гему. Придбана порфірія зустрічається при отруєннях (свинцем, барбітуратами та ін), гіповітамінозах (пелагра), перніціозної анемії, деяких хвороб печінки. Відкладення порфірину у шкірі викликають фотодинамічний ефект (еритема, дерматит).
Залізовмісний пігментутворюється також при розпаді міог-лобіну. Міосидерин виявляють в м'язовій тканині, що атрофується, але більшою мірою при дистрофії та воскоподібному некрозі її, пов'язаних з білом'язовою хворобою тварин, паралітичною міоглобінурією коней. При цьому міоглобінемія супроводжується розвитком міогемосидерозу органів, багатих на ретикулоендотеліальну тканину (селезінка, печінка, лімфовузли та ін.), виділенням розчиненого пігменту з сечею (міоглобінурія з червоним кольором сечі) та відкладенням його в епітеліальних клітинах сечових канальців.
При деяких отруєннях (нітритами та ін.) загальна пігментація пов'язана з утворенням метгемоглобіну (світло-коричневий колір крові). Гемоглобін може набувати зеленого або чорного забарвлення, якщо він з'єднується з сірководнем і утворюється сірчисте залізо (хибний меланоз). У свіжих трупів коней пігментацію відзначають у здухвинній кишці у вигляді плоских ділянок чорного кольору, що підносяться.
Протеїногенні (тирозинтриптофанові) пігментивключають меланін, андренохроми і пігмент ентерохромафінних клітин.
Меланін(від грец. melanos - чорний) утворюється в меланобластах -клітинах неврогенної природи базального шару епідермісу, волосяних цибулин, сітківки та райдужної оболонки, надаючи їм специфічний колір (чорний, бурий, жовтий, рудий). Це продукт природної полімеризації тирозину та триптофану, який синтезується в присутності вітаміну С у безбарвний промеланін, а під впливом тирозинази (допаоксидази) перетворюється на меланін. Пігмент не містить заліза та жиру, знебарвлюється перекисом водню та іншими сильними окислювачами, відновлює аміачний розчин нітрату срібла до металевого срібла, розчинний лише у лугах. У цитоплазмі меланобластів пігмент відкладається у вигляді зерен та глибок темно-коричневого кольору. Міграцію меланіну в організмі забезпечують макрофаги - меланофори, які через відсутність тирозинази не здатні до синтезу меланіну. На відміну від меланобластів вони не дають позитивну додаткову реакцію.
Порушення меланогенезу виявляються підвищеним утворенням меланіну, накопиченням їх у незвичайних місцях, зникненням чи відсутністю пігменту. Всі три види розладу обміну можуть бути придбаними або вродженими та мати поширений або місцевий характер.
Надмірне утворення меланіну в шкірі та відкладення його у внутрішніх органах називають загальним меланозом, який зустрічається головним чином у великої та дрібної рогатої худоби, особливо у телят та овець. Природа меланозу невідома, але вважають, що це процес кормового походження. Відзначають його у тварин, що випасалися на пасовищах із заболоченими та закисленими ґрунтами. Меланін відкладається в печінці, легенях (рис. 9) і на серозних покривах, рідше - в оболонках головного і спинного мозку, які набувають темно-коричневого або буро-чорного кольору. Зазвичай меланоз виявляється після забою тварин. Поширений меланоз з пігментацією шкіри та слизової оболонки ротової порожнини у бронзовий колір спостерігають у собак при аддісоновій хворобі у зв'язку з ураженням надниркових залоз. Посилена пігментація шкіри буває у сільськогосподарських тварин при хронічних хворобах, що супроводжуються виснаженням.
Місцева надмірна пігментаціяШкіра пов'язана з доброякісним або злоякісним розростанням меланобластів з утворенням меланом. Нерідко вони виникають у коней сірої масті та у собак. Джерела появи їх - родимі плями (naevus).
Внаслідок розпаду пігментних пухлин може розвинутися вторинний загальний меланоз.
Вроджене недостатнє утворення меланіну або його повна відсутність в організмі називається альбінізмом (albus – білий). Це пов'язане з рецесивним геном і відсутністю пігментоутворюючих тирозиназ. Його спостерігають у коней сірої масті, у великої рогатої худоби деяких порід (герефорди), овець, хутрових звірів, білих ведмедів, кроликів та ін. слабкість та схильність до захворювань. Ця хвороба у людей і тварин описується як Шедіак-Хігаші-синдром. Можливо і місцева вроджена депігментація шкіри (vitiligo). Набуті безпігментні плями, які називають лейкодермією (від грец. leukos — білий, derma — шкіра), утворюються після тривалих запалень та інших уражень шкіри (поранень, виразок, при випадковій хворобі коней та ін.).
До ліпідогенних пігментів, або ліпопігментів, належать ліпофусцин, цероїд та ліпохроми. До їх складу входять жирові та білкові речовини.
Ліпофусцин- гліколіпопротеїд, утворюється в клітинах в процесі аутооксидації фосфоліпідів. Під мікроскопом має вигляд зерен та глибок бурого кольору. Пігмент суданофілен, забарвлюється шарлахом у червоний колір, нерозчинний в органічних розчинниках та кислотах, частково розчинний у лугах, на відміну від меланіну при взаємодії з азотистокислим сріблом не чорніє. Ліпофусцин є нормальним компонентом клітини, що бере участь в окислювальних процесах.
Патологічну пігментацію ліпофусцином, особливо печінки, нирок, серцевого та скелетного м'язів, нервових клітин, спостерігають при виснажливих хворобах, наприклад, при вуглеводно-білковій недостатності у корів з високою продуктивністю, при атрофії паренхіматозних органів, у тому числі в старості (стареча атрофія). Макроскопічно в міру накопичення пігменту орган набуває бурого кольору (бура атрофія).
Пігменти гемофусцин, що виявляється в печінці у коней при інфекційному енцефаломієліті, і цероїд, освіта якого пов'язана з гіповітамінозом Е, за фізико-хімічними та біологічними властивостями ідентичні ліпофусцину.
Ліпохроми- пігменти, що надають жовте забарвлення жировій клітковині, корі надниркових залоз, жовтку яєць, сироватці крові і т. д. До ліпохром також відноситься лютеїн - пігмент жовтого тіла яєчників. Вони є ліпіди, у яких розчинені пофарбовані вуглеводні — каротиноїди і флавини. Утворення їх тісно пов'язане з білково-жировим метаболізмом та обміном рослинних пігментів. При обробці гістозрізів кислотами (наприклад, сірчаної) останні дають нестійке зеленувато-блакитне фарбування, під впливом окисних ферментів бліднуть, в ультрафіолетовому світлі мають зелену флюоресценцію, під впливом алкоголю випадають у кристали. Посилену пігментацію ліпохромами жирової клітковини відзначають при виснаженнях у зв'язку з конденсацією пігменту. При цьому клітковина набуває яскраво-жовтого забарвлення. Жовте фарбування та жовто-бурий колір кісток зустрічають при порушеннях ліпідно-вітамінного обміну (цукровий діабет та ін.), а також у місцях накопичення холестерину (в атероматозних бляшках та ксантомах).
Екзогенні пігментаціїпов'язані з надходженням в організм чужорідних барвників з зовнішнього середовища. Найбільш часто зустрічається відкладення в легких пилових частинок мінерального, рослинного або тваринного походження з розвитком пневмоконіозів (від грец. Рпеітоп - легке, conia - пил). Ці частки адсорбуються на слизових оболонках, впроваджуються в епітеліальні клітини, фагоцитуються макрофагами, проникають у лімфатичні судини та вузли, а також можуть заноситися до інших органів.
Серед цих захворювань важливе практичне значення мають антракоз легень, пов'язаний із відкладенням у них вугільного пилу. Найчастіше антракоз зустрічається коней та собак. Легкі при цьому набувають дифузного або строкатого аспідно-чорного або шиферного забарвлення. Значне відкладення вугільного пилу викликає запальні зміни, розвиток сполучної тканини та індурацію легких. З легких вугільні частинки поширюються в регіональні лімфатичні вузли, рідше - в селезінку та печінку. У великої рогатої худоби часто буває антракоз мезентеріальних лімфатичних вузлів при згодовуванні тварин запиленого корму. Відкладення в легких кремнезему, глинозему, глибок кварцу з утворенням білих вогнищ називається силікозом.
При тривалому лікуванні тварин препаратами срібла може розвинутись артроз. Солі срібла відкладаються в епітелії сечових канальців і в мезангіумі судинних клубочків, а також в ретикуло-ендотеліальних клітинах печінки та інших органів, тканини яких набувають сірого (сталевого) кольору. Певне забарвлення надають органам деякі лікарські (наприклад, метиленова синька, пікринова кислота) та барвники, що застосовуються при татуюванні тварин.
Порушення обміну нуклеопротеїдів.Нуклеопротеїди є сполуками білків з нуклеїновими кислотами — дезоксирибонуклеїновою (ДНК) і рибонуклеїновими (РНК). До порушень нуклеопротеїдного обміну відносять мочелікий діатез і сечокислі інфаркти.
Сечокислий діатез(Від грец. Diathesis - схильність) характеризується підвищеним утворенням та накопиченням сечової кислоти та її солей у крові (гіперурекемія) з подальшим відкладенням кристалів сечової кислоти та аморфного сечокислого натрію в різні тканини та органи. Найчастіше сечокислий діатез зустрічається у птахів, особливо з загону курячих, рідше - у ссавців (собаки та ін.).
Поява цього захворювання у птахів в умовах клітинного вмісту, у тому числі і у диких у зоопарку, пов'язують із рясним та тривалим білковим харчуванням продуктами тваринного (м'ясо, риба, м'ясо-кісткове та рибне борошно) та рослинного (концентровані корми) походження, особливо при нестачі зелених та інших вітамінних кормів (зокрема, вітаміну А). З внутрішніх чинників цьому сприяють захворювання нирок та печінки.
Локалізацію патологічних процесів у певних органах можна пояснити особливим фізико-хімічним та алергічним станом тканин, що затримують сечову кислоту та її солі.
При мікроскопічному дослідженні у місцях відкладення маси з променистими кристалами сечової кислоти та аморфних опадів її солей виявляють некротичні вогнища, навколо яких утворюється запальний інфільтрат з наявністю лейкоцитів, гістіоцитів та особливо характерних гігантських клітин. За ексудативно-клітинною реакцією наступають більш менш виражені проліферативні зміни, які супроводжуються утворенням грануляційної та фіброзної тканин з деформацією уражених органів.
Макроскопічні зміни характеризуються тим, що сечова кислота та сечокислий натрій випадають на серозних оболонках, нирках та інших внутрішніх органах, а також у суглобах кінцівок (хрящах, синовіях, сухожильних піхвах). Тому розрізняють вісцеральну, суглобову та змішану форми хвороби.
При вісцеральному сечокислому діатезі(Зустрічається тільки у птахів) сечова кислота та її солі у вигляді білих меловидних мас або дрібного кристалічного порошку відкладаються на серозних оболонках грудобрюшної порожнини, повітроносних мішків, нирок, печінки, селезінки, кишечника, серця та легень, а також інших органів. Під накладаннями, що легко знімаються, виявляється запалений серозний покрив. При тяжкій формі діатезу накладення стають гіпсовидними, серозні оболонки злипаються і зростаються. У внутрішніх органах, особливо в нирках, а також у печінці, підшлунковій залозі, серцевому та скелетному м'язах (м'язах ніг, крил), в ендокарді та ендотелії великих судин, під шкірою, у залозистому шлунку виявляють відкладення сечової кислоти та уратів у формі розсіяних точок. , плям, смуг або схильних до злиття біло-жовтих вузликів. У цьому уражені органи, особливо нирки, збільшуються обсягом.
Суглобова форма хвороби, або подагра(Від грец. Pous-нога, agrios - жорсткий), характеризується відкладенням сечової кислоти і уратів на синовіальних оболонках суглобів і сухожильних піхв, в капсулах суглобів і навколишніх тканинах. Найчастіше уражаються скакальні та пальцеві суглоби. Уражені суглоби збільшені, тверді, деформовані, з помірно щільними фіброзними вузлами - подагрическими шишками (tophi unci), в яких виявляють суху меловидную або вершкоподібну масу. При цьому в суглобовому хрящі можуть виникати некрози та виразки у вигляді виїмок (узурів), а навколо них запальна реакція зі скупченням гігантських клітин та розростанням сполучної тканини.
Сечокислі інфаркти нирок(infarcire - начиняти, нафаршувати) зустрічаються переважно у новонароджених. Сечова кислота і її солі відкладаються в гомогенній глікопротеїдній масі в просвіті прямих канальців, в апікальній частині залозистого епітелію і в стромі органу, в мозковому шарі і сосочках нирок, утворюючи білуваті, білувато-жовті або червонувато-жовті радіально смужки.
У просвіті прямих канальців та у проксимальному відділі нефрону вони зустрічаються у меншій кількості.
Сечокислі інфаркти з'являються у зв'язку з масовим розпадом еритроцитів, що містять ядро, при переході плода на режим зовнішнього дихання, з перебудовою харчування і обміну. При цьому у крові різко зростає концентрація сечової кислоти. Утворенню інфарктів, крім того, сприяє втрата новонародженої води. Як показали наші спостереження, конгломерати кристалів сечокислого амонію і пухка білкова маса, що їх пов'язує, можуть стати основою для розвитку сечокам'яної хвороби у молодих тварин, зокрема у норок.
Інкрустація мертвих мас.У дорослих тварин сечова кислота та її солі можуть просочувати мертві тканини та випадати в них в осад. Це відбувається у тканинах сечовивідних проток при зіткненні мертвої маси із сечею.
порушень обміну нуклеопротеїдів.При сечокислому діатезі порушуються функції уражених органів (нирки, печінка та ін.). Суглобова форма хвороби супроводжується деформацією, малою рухливістю та хворобливістю уражених суглобів. Гіперурекемія та гіперазотемія можуть бути причиною раптової смерті тварини. Відкладення сечової кислоти та уратів в органах викликають незворотні (некротичні) зміни уражених тканин.
Порушення обміну глікопротеїдів. Глікопротеїди – складні сполуки білка з полісахаридами, що містять гексози, гексозаміни та гексуронові кислоти. До них відносять муцини та мукоїди (про інші глікопротеїди див. «Вуглеводні дистрофії»).
Мучини складають основу слизу, що секретується епітелієм слизових оболонок та залоз. Слиз має вигляд напівпрозорої тягучої речовини, що випадає під впливом слабкої оцтової кислоти або алкоголю у вигляді тонкої сітки. До складу слизу входять нейтральні або кислі полісахариди - білкові комплекси, що містять гіалуронову та хондроїтинсерну кислоти (глікозаміноглікани), які надають слизу хромотропні або мета-хроматичні властивості. Тіонін і крезилвіолет забарвлюють слиз у червоний колір, а тканини - у синій або фіолетовий. Муцикармін надає їй червоного кольору, а толуїдиновий синій — бузково-рожевий.
Слизоутворення як патологічний процес має захисно-пристосувальне значення. Муцин захищає слизові оболонки від фізичних ушкоджень та подразнень хімічними речовинами. Слиз є носієм травних ферментів.
Мукоїди, або слизоподібні речовини (псевдомуцини), не однорідні за складом хімічні сполуки, що містять білок і глікозаміноглікани. Вони входять до складу різних тканин: кісток, хрящів, сухожиль, клапанів серця, стінок артерій та ін. В ембріональних тканинах борошна містяться у великій кількості, в тому числі в пупковому канатиці новонароджених. Вони мають загальні фізико-хімічні властивості зі слизом. Мукоїди мають лужну реакцію і на відміну від муцину не осаджуються спиртом або оцтовою кислотою.
Слизова дистрофія супроводжується накопиченням слизу та слизоподібних речовин у тканинах. Розрізняють два види її: клітинну (паренхіматозну) та позаклітинну (мезенхімальну).
Клітинна (паренхіматозна) слизова дистрофія- Порушення обміну глікопротеїдів у залозистому епітелії слизових оболонок, які проявляються гіперсекрецією слизу, зміною якісного складу її і загибеллю секретуючих клітин.
Слизова дистрофія частіше виникає при катаральних запальних процесах на слизових оболонках внаслідок прямої або непрямої (рефлекторної) дії різних патогенних подразників. Її відзначають при захворюваннях травних, дихальних та сечостатевих органів.
Роздратування слизових оболонок викликає розширення площі секреції та підвищення інтенсивності слизоутворення, а також зміна фізико-хімічних властивостей та складу самої слизу.
Гістологічнослизова дистрофія характеризується гіперсекрецією або надлишковим утворенням муцину в цитоплазмі епітеліальних (головним чином бокалоподібних) клітин, що вистилають слизові оболонки, підвищеним слизовим виділенням, загибеллю і десквамацією клітин, що секретують. Слиз може закривати вивідні протоки залоз і викликати утворення ретенційних кіст, чому сприяє здавлюванню їх сполучною тканиною, що розростається. При більш рідкісному поліпозному катарі, навпаки, спостерігають гіперплазію як залізистої, а й сполучної тканини.
Макроскопічнослизова оболонка набрякла, тьмяна, покрита товстим шаром слизу, при гострому запаленні органу вона гіперемована з крововиливами, а при хронічному - ущільнена через розрост сполучної тканини. Продукований у великій кількості слиз залежно від ступеня гідратації або дегідратації та кількості десквамованих клітин буває різної консистентності та в'язкості. Залежно від виду запалення органу до слизу домішується ексудат різного складу (серозний, гнійний, геморагічний).
Функціональне значенняі результат слизової дистрофії залежить від інтенсивності і тривалості процесу. Усунення патогенних чинників регенерація епітелію з допомогою камбіальних клітинних елементів може призвести до повного відновлення уражених органів. Тривалий поточний дистрофічний процес супроводжується загибеллю клітинних елементів епітелію, розростом сполучної тканини та атрофією залоз. У цих випадках відзначають різко виражену функціональну недостатність органу (наприклад, часткове випадання травної функції органів шлунково-кишкового тракту та при хронічному катарі з розвитком виснаження тощо).
Своєрідним різновидом порушення обміну глікопротеїдів є колоїдна дистрофія (від грец. Colla - клей), яка характеризується надлишковим утворенням і накопиченням колоїдної маси псевдомуцину в залозистих органах (щитовидні залози, нирки, надниркові залози, гіпофіз, яєчники, слиз). Фізіологічний прототип колоїду - секрет щитовидної залози. Зустрічається ця дистрофія при колоїдному зобі, пов'язаному з йодною недостатністю (ендемічне захворювання людей та тварин у певних геобіохімічних зонах).
Мікроскопічноспостерігаються гіперсекреція колоїду, накопичення його у фолікулах, атрофія залізистої тканини, розрив оболонок та злиття фолікулів з утворенням кіст. Новоутворені залізисті фолікули шляхом брунькування від попередніх також можуть зазнавати колоїдної дистрофії.
Макроскопічнощитовидна залоза, рідше інші залізисті органи збільшуються в обсязі, стають нерівними з поверхні, на розрізі в них виявляють кісти з тягучим вмістом клеєвидним від сірувато-жовтого до темно-коричневого кольору.
Колоїдна дистрофія викликає функціональну недостатність органу. При колоїдному зобі розвивається загальний слизовий набряк сполучної тканини (мікседема).
Позаклітинна (мезенхімальна) слизова дистрофія(Слиснення, слизовий метаморфоз) - патологічний процес, пов'язаний з накопиченням у сполучній тканині (волокнистої, жирової, хрящової та кісткової) хромотропних речовин.
Причинитканинної слизової дистрофії: виснаження та кахексія будь-якої етіології, наприклад при голодуванні, хронічних хворобах (туберкульоз, злоякісні пухлини та ін.) та дисфункції залоз внутрішньої секреції (колоїдний зоб та ін.). Сутність слизового метаморфозу полягає у вивільненні із зв'язку з білком хромотропної речовини (глікозаміногліканів) і накопиченні його в основному речовині сполучної тканини.
Гістологічнона відміну від борошна набухання відбувається розчинення колагенових волокон і заміщення їх слизоподібної масою. Клітинні елементи при цьому відокремлюються, набухають, набувають неправильної форми багатовідросткової або зірчастої, а також розчиняються.
Макроскопічноуражені тканини стають набряклими, в'ялими, студневидними, просоченими слизоподібною масою, що напівпросвічує.
Функціональне значення та результатцього процесу визначаються ступенем та місцем його розвитку. У початкових стадіях ослизнення усунення причини супроводжується відновленням структури, зовнішнього вигляду та функції ураженої тканини.
У міру розвитку процесу відбуваються повне розрідження та колікваційний некроз тканини з утворенням порожнин, заповнених слизоподібною масою.
Жирові дистрофії (ліпідози)
Жирові дистрофії (ліпідози) - морфологічні зміни тканин, пов'язані з порушенням обміну ліпідів.
Вільний жир у клітинах і тканинах має вигляд крапель, іноді кристалів (холестерин), розчинний в органічних розчинниках: спирт-ефір, хлороформ, нерозчинний у воді (на відміну від глікогену) і в оцтовій кислоті (на відміну від білків). Судан III і шарлах у заморожених гістозрізах, зафіксованих формаліном, забарвлюють його у червоний колір, судан IV та осмієва кислота – у чорний. Нільблаусульфат забарвлює жирні кислоти (ліпоїди) у темно-синій колір, нейтральний жир – у червоний. Відновлення жиром осмієвої кислоти з утворенням осміофільних включень дозволяє виявляти його при електронній мікроскопії. Електронно-мікроскопічно ліпідні включення зазвичай виявляють у вигляді вільнолежачих, не оточених мембраною крапель або кристалів (на відміну від секреторних жирових включень у залозистих органах, наприклад, у молочній залозі).
Порушення ліпідного обміну можуть бути клітинні, або паренхіматозні (порушення обміну цитоплазматичного жиру), позаклітинні або стромально-судинні (порушення обміну жиру в жировій клітковині), і змішані (системні ліпоїдози та ін.). За механізмом розвитку розрізняють: інфільтрацію, трансформацію, декомпозицію, тобто розпад клітинних білково-ліпідних комплексів, мембран та макромолекул, і змінений, або «збочений», синтез. У цьому змінюється як кількісний вміст жиру, а й його якісний склад із появою продуктів розпаду жиру.
Клітинні (паренхіматозні) жирові дистрофії- Порушення обміну цитоплазматичного жиру з накопиченням його в органах і тканинах, паренхімні клітини яких в нормі містять мало вільного жиру (печінка, нирки), не містять його взагалі (міокард, скелетний м'яз, нервова тканина та ін) або в них утворюється жир незвичайного хімічного складу внаслідок патологічного синтезу.
Причиниданої дистрофії: загальне ожиріння, вуглеводна та білкова недостатність, дефіцит ліпотропних факторів, наприклад холіну, метіоніну, інших глюкопластичних амінокислот, вітаміну В12 та ін. (Аліпотропна, або проста, жирова дистрофія). Жирова дистрофія часто зустрічається у поєднанні із зернистою дистрофією при хворобах обміну речовин, серцево-судинної системи та кровотворних органів (анемії, розлади кровообігу), а також при багатьох інфекціях, інтоксикаціях та отруєннях різними отрутами, наприклад фосфором, миш'яком, чотирихлористим вугіллям. (Дистрофічне ожиріння).
Патогенезжирової дистрофії пов'язаний з інфільтрацією, тобто з відкладенням у клітинах жиру, що приноситься зі струмом лімфи та крові зі шлунково-кишкового тракту, мобілізацією жирних кислот із жирових депо, а також з осередків розпаду жирової тканини. Можливий підвищений синтез або трансформація жиру з вуглеводів і білків, особливо при надмірному надходженні їх (просте ожиріння).
Найчастіше жирова дистрофія розвивається внаслідок зниження окисних процесів та уповільненої асиміляції жиру в патологічно змінених клітинах (дистрофічне ожиріння).
Механізм такого дистрофічного ожиріння пов'язаний з порушенням окисних процесів у циклі Кребса - Ембден - Мейергофа (у мітохондріях) через нестачу кисню або легко окислюваних субстратів (вуглеводів і глюкогенних амінокислот), що сприяють окисленню жирних кислот і кетонових тіл, або з блоком токсичних речовин на ферменти окисного фосфорилювання (фермен-топатія).
Поряд з екзогенним ожирінням джерелом дистрофічного ожиріння є ендогенний жир клітини, що входить до складу мембран, білково-жирових комплексних сполук, які також під дією вищевказаних причин (гіпоксія, інфекція, інтоксикація та ін) можуть піддаватися більш-менш вираженій декомпозиції, або ліпофанерозу ( від грец.lipos - жир, phaneros - видимий). В основі деструкції лежать ферментативні (гідролітичні) та фізико-хімічні процеси, наприклад, дегідратація.
У розвитку жирової дистрофії поряд із загальним її механізмом (інфільтрацією, трансформацією, декомпозицією) важливу роль відіграють структурно-функціональні особливості органів і тканин.
У печінці при жировій інфільтрації в цитоплазмі гепатоцитів (в її периваскулярній зоні) спочатку з'являються окремі дрібні краплі жиру (дрібнокрапельне периваскулярне ожиріння), які в міру накопичення переміщаються до центру (центральне ожиріння) і зливаються в більші краплі (круп) нарешті, в одну велику краплю жиру; остання відтісняє ядро і цитоплазму, що атрофується, до периферії клітини, надаючи їй перстоподібну форму (рис. 12), властиву клітинам жирової тканини. Жирова інфільтрація може бути перилобулярною, центролобулярною або дифузною.
Електронно-мікроскопічно та гістохімічноу печінці при жировій інфільтрації різного ступеня (легка, середня та важка) встановлені набухання та зменшення числа мітохондрій, розпад полісом та рибосом гепатоцитів, зменшення або повне зникнення гранул глікогену, зниження активності окисно-відновних ферментів, поява крапель жиру в зоні більш менш вираженим накопиченням його в гіалоплазмі. При жировій декомпозиції йдеться про ліпофанероз білково-жирових комплексних сполук (ліпопротеїдів), що містяться в зоні цитоплазматичної мережі з накопиченням ліпосом, та про розпад органел. Мітохондрії піддаються жировому метаморфозу, при цьому утворюються цитолизосоми з підвищеною активністю гідролітичних ферментів (кислої фосфатази), а потім і ліпофусцину (А. В. Жаров, 1975).
При осередковій жировій декомпозиції з розпадом ядер виникають ділянки жирового некрозу, наприклад в печінці, в осередках розм'якшення головного мозку та ін. та зернисті кулі. Клітини, що фагоцитують холестерин, набувають пластинчастої форми. У зв'язку з тим, що скупчення таких клітин макроскопічно мають вигляд жовтих плям, їх назвали ксантомними (від грец. xanthos — жовтий).
Зовнішній вигляд печінки при жировій дистрофії суттєво змінюється. Жирова інфільтрація перилобулярного типу разом із гострої застійної гіперемією надає їй мускатний малюнок. При вираженій жировій дистрофії печінка збільшена, жовто-коричневого кольору, сальна, в'яла, малюнок часток згладжений, на поверхні ножа при розрізі залишається сальний наліт. Крайні форми дистрофічного ожиріння печінки настільки знижують густину органу, що його шматочки можуть плавати у воді, як це спостерігається у молочних корів при кетозі.
У нирках нейтральний жир як фізіологічне явище зустрічається в епітелії вставних відділів канальців, петель Генле та збиральних трубочок. Макроскопічно при жировій дистрофії нирки збільшуються, набувають сіро-жовтого кольору, малюнок шарів згладжений, поверхня розрізу органу жирна, липка.
Жирова дистрофія міокарда проявляється як жирова інфільтрація та декомпозція. Жирова інфільтрація характеризується відкладенням дрібних крапель жиру в зоні капілярної та венозної мереж у зв'язку з гіпоксією (дрібнокрапельне ожиріння). У початковій стадії розвитку її дрібні крапельки жиру орієнтовані по ходу міофібрил, а потім поперечна смугастість зникає, саркосоми набухають, саркоплазматична мережа розширюється, рибосоми та глікоген розпадаються. При жировій декомпозиції утворення жиру пов'язане із розпадом органел. Жир може повністю заміщати саркоплазму волокон, що розпалися (міоліз). Макроскопічно такі ділянки виявляються у вигляді сірувато-жовтих смужок, що надають міокарду малюнок тигрової шкіри (тигрове серце).
Функціональне значенняжирової дистрофії паренхіматозних органів та спеціалізованих елементів інших тканин випливає з того, що функції органів при цьому знижуються, порушуються або випадають. При збереженні ядерного апарату клітин та частини органел цитоплазми жирова дистрофія оборотна. Посилення жирового некробіозу та некрозу ядер паренхімних клітин печінки, міокарда, нирок та інших органів супроводжується смертельним наслідком.
Позаклітинні (стромально-судинні) жирові дистрофії- Порушення обміну нейтрального жиру і жирних кислот у жировій клітковині, холестерину з його естерами. У патологічних умовах порушення обміну нейтрального жиру в жировій клітковині проявляються в виснаженні та ожирінні організму.
Виснаження (кахексія)- загальне зменшення кількості жиру в жировій клітковині з більш менш повною втратою вільного жиру в органах.
Причини:голодування тварин (аліментарна дистрофія), а також хронічні, виснажливі організм, інфекційні (туберкульоз), інвазійні (гельмінтози) та незаразні (гастроентерити, бронхопневмонії, пухлини, гормональні та обмінні розлади та ін.) хвороби.
При мікроскопіїжирової тканини виявляють зморщені клітини, а основний субстанції — скупчення серозної рідини чи слизеподобного речовини. Більш-менш виражені атрофічні процеси (з накопиченням ліпофусцину) знаходять і в паренхіматозних органах.
Макроскопічножирова клітковина втрачає жир, зменшується в обсязі, стає в'ялою, вологою внаслідок просочування серозною рідиною (серозна атрофія жиру), надалі розвивається ослизнення тканини (слизовий метаморфоз), вона набуває студнеподібного вигляду і жовтувато-сірого кольору.
Функціональне значення та результатвиснаження залежать від причини, що його викликала, можливості її усунення та ступеня патоморфологічних змін. Початкове і навіть клінічно виражене виснаження може бути оборотним.
Показником крайнього виснаження організму з неблагополучним результатом є серозна атрофія епікарда, бура атрофія печінки та міокарда. У старих тварин, особливо у великої рогатої худоби та коней, незворотна атрофія жиру може супроводжуватися деяким ущільненням клітковини внаслідок розросту сполучної тканини та темно-жовтого забарвлення її у зв'язку з конденсацією ліпохромів.
Регіонарне, або місцеве, зменшення кількості жиру в жировій клітковині називають ліподистрофією, яку виявляють при ендокринних захворюваннях (рецидивуючий панікуліт, що не нагноюється і ін.) і ліпогранулематоз. Сутність ліпогранулематозу полягає в осередковій деструкції жирової тканини з утворенням окисленого жиру, жирових кіст або запальних гранульом. Виникнення таких вогнищ пов'язане з травмою, деякими інфекційними хворобами (наприклад, стрептококозом) або з підшкірною ін'єкцією лікарських речовин.
Антиподом виснаження є загальне ожиріння із значним збільшенням жиру в жировій клітковині та відкладенням його в незвичайних місцях.
Причини:екзогенні фактори внаслідок перегодівлі тварин в умовах недостатньої рухливості та дефіциту кисню (аліментарне ожиріння) та ендогенні – у зв'язку з різними захворюваннями нервової (переважно у людей) та ендокринної систем. Аліментарне ожиріння при висококалорійних раціонах спостерігають у свиней, молочних корів наприкінці лактації та в сухостійний період, у овець, птахів та м'ясоїдних. Ендокринні розлади, що супроводжуються ожирінням, зустрічаються у тварин при гіпофункції яєчників (наприклад, у корів, м'ясоїдних та ін), а також інших залоз внутрішньої секреції.
Мікроскопічножирові відкладення виявляють поза жирової клітковини з утворенням нових жирових депо та у внутрішніх органах. При цьому паренхіматозні елементи атрофуються і заміщаються тією чи іншою мірою жировою тканиною. Наприклад, в інтерстиції вимені корів утворюється жирова клітковина, яка витісняє секреторну тканину. Ожиріння епікарда та сполучнотканинного каркасу серця супроводжується атрофією м'язових волокон.
Макроскопічнозагальне ожиріння проявляється у більш-менш багатих відкладеннях переважно нейтрального жиру не тільки в підшкірній клітковині, сальнику, брижі, під очеревиною, в середостінні, в епікарді, але і в сполучній тканині таких органів, де вільний жир в нормі буває в невеликих кількостях або зовсім Відсутнє. Наприклад, у міру накопичення подепікар-діального жиру у вигляді суцільного жирового прошарку відкладення його відбувається і в строму органа. У таких випадках серцевий м'яз, особливо правого відділу серця, набуває в'ялої консистенції, на поверхні розрізу міокарда і під епікардом виявляють жовтувато-білі смужки жирової тканини.
Загальне ожиріннявідноситься до оборотних процесів, за винятком випадків, обумовлених важким ураженням залоз. Особливе клінічне значення має залучення до процесу серця, яке проявляється функціональною недостатністю (міокардо-зом). Загальне ожиріння - одна з передумов розвитку кетозу, безпліддя та інших ускладнень, що є причиною передчасного вибракування або вимушеного забою таких тварин.
Місцеве надмірне накопичення жиру, або ліпоматоз, в основі якого лежить вакатне розростання сполучної тканини, що зустрічається при атрофії органів (у фізіологічних умовах при атрофії зобної залози, у патологічних - нирки, окремих лімфовузлів, ділянок скелетних м'язів та інших органів).
Порушення обміну холестерину та його естерівспостерігають при серцево-судинних захворюваннях типу артеріо- та атеросклерозу (від грец. athere - кашкоподібна маса, scleros - ущільнення).
Сучасні гістохімічні та електронно-мікроскопічні дослідженняпоказали, що інфільтративному та резорбтивному ожирінню судинної стінки (гіперхолестеринемії та ліпемії) передує передхолестеринова стадія хвороби, пов'язана з порушенням обміну глікозами-ногліканів та глікопротеїдів, плазморагією, мукоїдним та фібриноїдним. 965). При цьому через підвищену судинну проникність в інтимі артерій накопичуються не тільки холестерин та його естери (Н. Н. Анічков, 1953), але також білки плазми крові: альбуміни, глобуліни, фібриноген, b-ліпопротеїди, а у тварин – переважно жир (А. Ф. Ткаченка, 1965). Все це супроводжується дистрофією та некрозом судинної стінки з утворенням макроскопічно виражених атероматозих бляшок білково-жирового детриту, розростом сполучної тканини та її гіалінозом із звуженням просвіту судин. В атероматоз-их бляшках зазвичай випадають солі кальцію на кшталт дистрофічого звапніння або на місці їх виникають виразкові дефекти можливими несприятливими наслідками (кровотеча, тромбоз і т. д.).
Вуглеводні дистрофії
Вуглеводними дистрофіями називають зміни складу та кількості вуглеводів у тканинах, обумовлені порушеннями їх всмоктування, синтезу та розпаду.
Більшість вуглеводів перебуває у складі комплексних сполук клітин та тканин. Гістохімічно виділяють полісахариди реакцією з Шифф-йодистою кислотою (ШІК- або PAS-реакцією Мак-Мануса). З огляду на те, що вуглеводи легко розчиняються у воді, для їх виявлення використовують спиртові фіксатори (фіксатор Шабадаша та ін.). У ШІК-реакції після окислення полісахаридів йодистою кислотою вивільняються альдегідні групи, які дають з фуксином Шиффа (фуксинсернистою кислотою) сполуки червоного кольору. За методом Беста глікоген забарвлюється у червоний колір.
У патології вуглеводного обміну розрізняють зменшення або збільшення глікогену в клітинах, а також патологічний синтез та відкладення його в органах та тканинах, у яких він у нормі не виявляється.
Причини: різко виражені зменшення кількості глікогену в печінці, скелетних м'язах та міокарді, що спостерігаються при гострому та хронічному голодуванні, гіпоксії, лихоманці, переохолодженні, а також при екзогенних та ендогенних інтоксикаціях та інфекціях. Дефіцит глікогену часто спостерігається при патології залоз внутрішньої секреції, що регулюють його обмін. Зменшення кількості глікогену встановлено за базедової хвороби у зв'язку з підвищенням інтенсивності основного обміну. Експериментально у жуйних це відтворюється при ін'єкціях тиреотропного гормону гіпофізу та тироксину з розвитком індукованого кетозу.
Мікроскопічноу тварин, особливо жуйних, вуглеводна недостатність із зменшенням або зникнення запасного глікогену з печінки та м'язової тканини часто поєднується із зернистою
дистрофією, мобілізацією жиру з підвищеним утворенням кетонових тіл та жировою інфільтрацією паренхіматозних органів, особливо печінки, нирок та міокарда (А. В.Жаров, 1975). Однак глікоген, пов'язаний із білками, повністю не зникає з клітин навіть при повному голодуванні. При цьому відзначаються патологічний синтез глікогену та відкладення його в нирках, епітелії вузького сегмента петлі Генле.
ПорушенняВуглеводні обміни яскраво виражені при цукровому діабеті (diabetus melitus). Сутність його полягає в недостатньому виробленні b-клітинами острівців Лангерганса гліколітичного гормону інсуліну з розвитком вуглеводної дистрофії, гіперглікемії, глюкозурії, поліурії, а нерідко й ускладнень кетозом та ангіопатіями. Цукровий діабет має панкреатичне (ураження інсулярного апарату) та позапанкреатичне (ураження вуглеводного центру, гіперфункція передньої частки гіпофіза та ін.) походження. Він часто зустрічається у людей. Хворіють собаки, рідше коні та велика рогата худоба. Експериментальний аллоксановий діабет (після введення алоксану або уреїду мезооксалєвої кислоти) можна викликати у щурів, кроликів, собак, мавп.
Гістологічнопри цукровому діабеті поряд з порушенням обміну глікогену в печінці і скелетних м'язах відзначають інфільтрацію глікогеном судинної тканини (діабетична ангіопатія), епітелію ниркових канальців (звивистих і петель Генле), стро-ми і судинних клубочків нирок з розвитком інтерка. При цьому іноді глікоген виділяється і у просвіт канальців.
Макроскопічнооргани при вуглеводній дистрофії немає характерних змін.
Клінічновідзначають функціональні розлади (пригнічення, серцева слабкість та задишка), пов'язані з енергетичною недостатністю. Причому ці зміни спочатку мають оборотний характер. Однак на основі вуглеводної дистрофії часто порушуються білковий та жировий обміни, розвиваються білкова та жирова дистрофії, які можуть супроводжуватися омертвінням клітин та несприятливим результатом.
Збільшення кількості глікогену у клітинах організму та його патологічні відкладення називаються глікогенозом.
Надмірний вміст глікогену спостерігають при анемії, лейкозах, у лейкоцитах і сполучнотканинних клітинах у запалених вогнищах, по периферії гострих інфарктів або туберкульозних вогнищ. Глікоген накопичується у відгодівельних тварин, особливо при гіпофункції щитовидної залози, спричиненої тиреостатиками (хлорнокислий амоній та ін.). Глікогенова інфільтрація зустрічається у тканинних елементах деяких пухлин (міом, сарком, карцином, невром та ін.). Особливо яскраво виражена патологічна інфільтрація клітин та тканин глікогеном відзначається у людей при хворобах, генетично обумовлених недостатністю ферментів глюкозо-6-глікозидази та ін.
Гістологічнопри цих хворобах відзначають надмірне накопичення глікогену в печінці (гепатоцити «нафаршировані» глікогеном), серце, нирках, скелетних м'язах, стінці судин та ін.
Макроскопічнонадмірне відкладення глікогену характерних ознак немає.
Клінічноглікогенози супроводжуються серцевою та дихальною недостатністю, від яких і настає смерть (Т. Є. Івановська, 1989). У тварин ці хвороби вивчені недостатньо.
Мінеральні дистрофії
Гістопрепарати.
| Текст поки що не знайдено. | |
| Інфільтраційне ожиріння, атрофія міокарда |
|
| Спостерігається стоншення м'язових волокон і втрата ними поздовжньої смугастість, поперечна смугастість відрізняється але не у всіх ділянках. Внаслідок зменшення обсягу м'язових волокон ядра їх лежать ближче одне до одного, тому кількість ядер у зору здається збільшеним. У різко виражених випадках змінюються форма і обсяг ядер (витягнуті в довжину, темні, а так само зморщені ядра). У сакроплазмі по полюсах ядер відкладається ліпофусцин у вигляді дрібних бурих зернят. З розвитком атрофії кількість пігменту збільшується, і він починає відкладатися в саркоплазмі по всьому волокну. Самі волокна при цьому можуть повністю атрофуватися і зазнати розпаду, а на їх місці залишаються купки пігменту. |  |
| Зерниста дистрофія з некробіозом гепатоцитів |
|
| Відзначають нерівномірне збільшення обсягу епітеліальних клітин та м'язових волокон, що здавлюють капіляри, набухання та помутніння цитоплазми, згладженість та зникнення тонкої структури (щіткової облямівки залозистого епітелію, і т. д.), поява та накопичення в цитоплазмі дрібної ациди. У цьому межі клітин та обриси ядер помітні важко. Іноді цитоплазма набуває пінистого вигляду, деякі клітини відокремлюються від базальної мембрани та одна від одної (дискомплексація). |  |
| Амілоїдна дистрофія |
|
| При загальному типовому амілоїдозі, найбільш поширеному у сільськогосподарських тварин, амілоїд випадає по ходу ретикулярних волокон судинних та залозистих мембран та в периретикулярні простори паренхіматозних органів (периретикулярний або паренхіматозний амілоїдоз). Уражаються печінка, селезінка, нирки, рідше надниркові залози, гіпофіз, власна оболонка залоз кишечника, інтима капілярів та артеріол. У сполучнотканинних клітинах накопичуються преамілоїдні фібрили, зникають рибосоми, гіпертрофуються мітохондрії (гігантські мітохондрії), а також пластинчастий комплекс Гольджі. Накопичення амілоїду в тканині супроводжується атрофією та загибеллю паренхіматозних елементів органу. |
 |
| Відбувається вилуговування солей кальцію та часткове розсмоктування вже сформованих кісток. Резорбція кісткової тканини в гаверсових каналах та інших місцях здійснюється ферментативним шляхом за участю остеокластів з утворенням порожнин, або лакун (лакунарна резорбція). Різне поєднання резорбції кісткової тканини, зниженого синтезу нових кісткових структур та демінералізації призводить до розвитку в одних випадках переважно остеопорозу, особливо при гормональній остеодистрофії, в інших — остеомаляції та остеофіброзу із заміщенням атрофічної кісткової тканини остеоїдної, хрякової. |  |
| У сильно розтягнутих фолікулах і особливо у великих порожнинах залозистий епітелій, видається низьким кубічним, сплощеним, що вказує на його атрофію. Цитоплазма клітин грубо зерниста, ядро піктонічне, межі клітин згладжені. Зустрічаються фолікули, у яких епітелій зберіг форму, але збільшений обсягом. У цитоплазмі таких клітин виявляють у великій кількості дрібні блискучі зерна колоїду, що іноді заповнюють цитоплазму і відтісняють ядро до переферії клітини. спостерігається також відшарування клітин від стінок фолікула. Одночасно з атрофічними процесами відзначають гіперплазію залізистого епітелію та формування нових фолікулів. |  |
Альтерація(Лат. alteratio - зміна) або ушкодження - це структурні зміни клітин і тканин організму, що виникають під дією екзогенних та ендогенних факторів. При цьому в тканинах та органах порушуються обмін речовин, функція та життєдіяльність. Причинами альтерації можуть бути порушення кровообігу, фізичні агенти та хімічні речовини, інфекційні агенти, імунопатологічні реакції, генетичні фактори та дисбаланс необхідних клітині речовин (зазвичай внаслідок порушення харчування). Ступінь пошкодження клітин та тканин залежить від виду та тривалості дії патогенного фактора, від морфо-функціональних особливостей макроорганізму. Альтерація може виникати на ультраструктурному, клітинному, тканинному та органному рівнях.
У патогенезі ушкодження клітин та тканин велике значення відіграють гіпоксичні фактори, пов'язані з недостатнім надходженням кисню до тканин (наприклад, при утрудненні притоку артеріальної крові). При гіпоксії
припиняється окисне фосфорелювання та зменшується синтез АТФ, активується гліколіз. Відбувається гостре набухання (набряк) клітини внаслідок порушення осмотичного тиску і накопичення в цитоплазмі метаболітів (фосфати, лактат і т.д.), набухають цистерни ендоплазматичної мережі, розширюються мітохондрії, ушкоджуються плазматичні мембрани, у тому числі і мембран їх ферментів та розщепленню компонентів клітини.
Пошкодження клітини може викликатись вільними радикалами кисню, хімічними факторами, іонізуючим випромінюванням; розвивається при старінні клітин. При цьому відбувається перекисне окиснення ліпідів мембран, що може призвести до руйнування плазматичної мембрани та органел. Значну роль також відіграє окисне перетворення білків, яке посилює руйнування ключових ферментів за допомогою нейтральних протеаз, та ушкодження вільними радикалами ДНК клітини.
Пошкодження можуть викликати хімічні речовини, які можуть діяти безпосередньо на молекули та органели клітини (наприклад, водорозчинні сполуки хлориду ртуті).
Альтерація представлена двома патологічними процесами – дистрофією та некрозом, які можуть бути послідовними стадіями.
Дистрофія(грец. dys - приставка, що позначає розлад і trophe - харчування) - складний патологічний процес, в основі якого лежать метаболічні порушення, що призводять до структурних змін. У морфологічному відношенні при дистрофії клітин і тканин з'являються речовини, яких в нормі немає або міститься мало, або ж з клітин і тканин зникають властиві для них речовини.
В основі дистрофії лежить порушення комплексу механізмів, що забезпечують обмін речовин і збереження структур клітин та тканин, таких як регулююча функція нервової та ендокринної систем, розлади кровопостачання та лімфообігу, а також розлади ауторегуляції клітини, що веде до порушення ферментативних процесів у клітині.
Вроджені порушення ферментних систем (ферментопатії) є причиною великої групи спадкових захворювань. тезаурізмозів (хвороб накопичення), при яких через відсутність будь-якого ферменту або його неправильної структури блокується будь-яка ланка метаболізму, і в клітинах з'являються аномальні речовини.
Розрізняють такі морфогенетичні механізми, що ведуть до розвитку дистрофії: інфільтрація декомпозиція (фанероз), збочений синтез, трансформація.
Інфільтрація - це надлишкове проникнення продуктів обміну з крові та лімфи в клітини або міжклітинна речовина з подальшим їх накопиченням. Типовим прикладом інфільтрації є інфільтрація внутрішньої оболонки стінки аорти (інтими) та великих судин холестерином та його ефірами, що є основою такого захворювання, як атеросклероз.
Декомпозиція або фанероз - це розпад ультраструктур клітин і міжклітинної речовини, що веде до порушення тканинного або клітинного метаболізму та накопичення продуктів порушеного обміну в тканині або клітині, наприклад, при дифтерійній інтоксикації виникає жирова дистрофія кардіоміоцитів. Збочений синтез – це синтез речовин у клітинах та тканинах, які не зустрічаються в нормі. Прикладом може бути амілоїдоз, зокрема при мієломній хворобі, коли В-лімфоцити, які в нормі продукують антитіла, при пухлинній трансформації продукують білки зі зміненою структурою.
Трансформація - Освіта продуктів одного виду обміну замість продуктів іншого виду обміну. Наприклад, трансформація компонентів жирів та вуглеводів у білки.
Класифікація дистрофії.
Залежно від того, в яких структурах переважно локалізуються дистрофічні зміни, їх ділять на паренхіматозні, стромально-судинні (мезенхімальні), змішані.
Залежно від переважання порушень тієї чи іншої видів обміну виділяють білкові, жирові, вуглеводні, мінеральні дистрофії, змішані.
Також розрізняють набуті та спадкові дистрофії, і залежно від поширеності – загальні та місцеві.
Паренхіматозні дистрофії
При цих дистрофіях речовини накопичуються в клітинах паренхіми різних органів, таких як міокардіоцити, гепатоцити, нейрони, клітини ниркових канальців, надниркових залоз. За видом порушеного обміну ці дистрофії ділять на білкові (диспротеїнози), жирові (ліпідози) та вуглеводні.
Паренхіматозні білкові дистрофії (диспротеїнози) характеризуються порушенням обміну цитоплазматичних білків, що у вільному чи пов'язаному стані . До них відносяться зерниста, гідропічна, гіаліново-крапельна та рогова дистрофії.
Зерниста дистрофія .
Причинами зернистої дистрофії можуть бути розлади кровообігу, інфекції, інтоксикації та інші фактори, що ведуть до зниження інтенсивності окисно-відновних процесів клітини. Органи при цьому процесі збільшені, в'ялі, на розрізі тьмяні, каламутні. Це дає підставу називати зернисту дистрофію каламутним (тьмяним) набуханням органів.
Мікроскопічно клітини збільшуються, цитоплазма їхня каламутна, багата на білкові гранули. Електронно-мікроскопічно при зернистій дистрофії відзначається збільшення кількості (гіперплазія) та набухання органел клітини, які світлооптично виглядають як білкові гранули. Така гіперплазія ультраструктур клітин в даний час розглядається як виражена функціональна напруга органів на різні дії.
Вихід зернистої дистрофії різний. У більшості випадків вона оборотна, але якщо причини, що її викликали, не усунуті, можливий її перехід у гіаліново-краплинну, гідропічну або жирову дистрофію.
Функція уражених органів може бути ослаблена.
Гіаліново-крапельна дистрофія .
Є важчим видом дистрофії. Найчастіше вона розвивається у нирках, печінці, рідше у міокарді.
При цьому вигляді дистрофії в цитоплазмі клітин з'являються великі краплі білка, що часто зливаються між собою і в результаті коагуляції білка, що нагадують основну речовину гіалінового хряща.
Зовнішній вигляд органів зазвичай не змінено або залежить від захворювань, при яких виникає цей диспротеїноз.
У нирках така дистрофія розвивається у тих випадках, коли через нирковий фільтр у сечу проникає велика кількість білка. При цьому накопичення білкових включень у цитоплазмі та її деструкція обумовлені неспроможністю лізосомальної реабсорбції білка епітелію ниркових канальців за умов підвищеної проникності гломерулярного фільтра (при нефротичному синдромі).
У печінці гіаліново-краплинна дистрофія частіше розвивається шляхом збоченого синтезу. Найчастіше вона розвивається при алкогольному ураженні, коли у цитоплазмі гепатоцитів з'являються краплі гіаліноподібного білка, які називаються тільцями Меллори (або алкогольним гіаліном) і є морфологічною ознакою алкогольного гепатиту. Іноді гіаліново-краплинна дистрофія гепатоцитів може спостерігатися при біліарних цирозах та хворобі Вільсона-Коновалова. Алкогольний гіалін може надавати цитолітичну дію на гепатоцити та стимулювати синтез колагену, що визначає хронічний прогресуючий перебіг алкогольного гепатиту та сприяє розвитку цирозу печінки.
Функція органів при гіаліново-крапельній дистрофії зазвичай значно страждає. Результатом цієї дистрофії може бути коагуляційний некроз (загибель) клітини.
Гідропічна (водяна) дистрофія
Ця дистрофія характеризується появою у клітинах вакуолей, наповнених цитоплазматичною рідиною. Виникає вона при порушеннях водно-електролітного та білкового обміну, що буває при інтоксикаціях різного генезу, а також при інфекціях, особливо вірусних. Так, вона розвивається в клітинах епідермісу та слизових оболонок при герпетичній інфекції, вітряній та натуральній віспі; у гепатоцитах – при вірусних гепатитах; у нейронах головного мозку та їх відростках – при спонгіоформних енцефалопатіях (у тому числі хвороби Крейтцфельда-Якоба), що викликаються інфекційною білковою молекулою – пріоном. Вакуольні дистрофії можуть піддаватися осьові циліндри нервових волокон провідної системи спинного мозку при аміотрофічному лейкоспонгіозі і мієлінові оболонки нервових волокон при ВІЛ-інфекції (вакуолярна мієлопатія).
Зовнішній вигляд органів зазвичай не змінений, за винятком шкіри при герпетичній інфекції та віспі, коли з'являються бульбашки (везикули), заповнені серозною рідиною.
При мікроскопічному дослідженні змінені клітини збільшені обсягом, цитоплазма їх заповнена вакуолями різних розмірів, а ядро зміщується на периферію клітини. При прогресуванні процесу вакуолі зливаються один з одним і клітина перетворюється на бульбашку, що представляє собою одну велику вакуолю і зміщене пухирцеподібне ядро ( балонна дистрофія)
У нирках гідропічна дистрофія епітелію звивистих канальців може бути обумовлена недостатністю системи реабсорбції базального лабіринту, який переповнюється водою, що проникає в клітину, і, "піднімаючись" до щіткової облямівки, руйнує мембрани і утворює балонні структури.
Гідропічна дистрофія гепатоцитів найчастіше є наслідком впливу вірусу гепатиту В чи токсичних речовин. При дії вірусу цей диспротеїноз виникає шляхом збоченого синтезу, підпорядкованого репродукції вірусу, а при дії токсичних речовин зумовлена недостатністю системи детоксикації.
Рогова дистрофія
Цей стан характеризується надлишковим утворенням рогової речовини в багатошаровому плоскому ороговіючому епітелії (шкіра), або ороговенням багатошарового плоского неороговевального епітелію (стравохід, шийка матки).
Рогова дистрофія може виникати при порушеннях розвитку шкіри, хронічних порушеннях кровообігу, хронічному запаленні, інфекціях, авітамінозах, пухлинах та ін.
Рогова дистрофія шкіри може бути загальною. іхтіоз– спадкове захворювання, що проявляється надмірною кератинізацією – надмірним лускатим лущенням), та місцевою ( гіперкератоз).
Ороговіння багатошарового плоского неороговеючого епітелію, наприклад, у порожнині рота або в шийці матки, макроскопічно виглядає у вигляді білої плями ( лейкоплакія) .
До спадкових паренхіматозних диспротеїнозів, які обумовлені порушенням внутрішньоклітинного метаболізму амінокислот, відносять: цистиноз, тирозиноз та фенілпіровиноградну олігофренію (фенілкетонурію). При цих захворюваннях найчастіше уражаються печінка, нирки, кістковий мозок, селезінка та нервова система.
Паренхіматозні жирові дистрофії (ліпідоз ).
Ці дистрофії характеризуються порушенням обміну жирів, переважно нейтральних, у цитоплазмі клітин. Переважання того чи іншого механізму виникнення ліпідозу залежить як від його причини, так і від структурно-функціональних особливостей органу. Найчастіше розвиток жирової дистрофії пов'язаний з гіпоксією, яка виникає при хронічних захворюваннях серцево-судинної системи, органів дихання, хворобах системи крові, а також з дією токсичних речовин (різні інфекції та інтоксикації) та збільшенням рівня жирних кислот у плазмі крові (загальне ожиріння ).
Зазвичай жирова дистрофія розвивається у серці, печінці, нирках.
Жирова дистрофія міокарда може виникнути найчастіше є результатом гіпоксії чи інтоксикації (дифтерійна, алкогольна, отруєння фосфором, миш'яком тощо.). І в тому і в іншому випадку розвивається пошкодження мітохондрій та зниження інтенсивності окислення жирних кислот.
У м'язових клітинах з'являються дрібні жирові краплі (пилоподібне ожиріння), які при наростанні змін (дрібнокрапельне ожиріння) замінюють цитоплазму. Процес носить осередковий характер і спостерігається в групах м'язових клітин по ходу венозного коліна капілярів і дрібних вен, чим і пояснюється своєрідний зовнішній вигляд серця: з боку ендокарда видно жовті поперечні смуги (“тигрове” серце). Міокард при цьому в'ялий, розміри серця можуть бути збільшені, а розширені порожнини.
Результат паренхіматозної жирової дистрофії міокарда залежить від її вираженості. Початкові зміни оборотні, глибокі ведуть до вираженого порушення функції.
Жирова паренхіматозна дистрофія печінки зустрічається найчастіше.
Вона може виникнути при підвищенні рівня жирних кислот у плазмі (цукровий діабет, загальне ожиріння), в результаті декомпозиції (при інфекціях та інтоксикаціях), трансформації (при хронічному алкоголізмі), при порушенні харчування внаслідок нестачі білка в їжі, при генетичних дефектах ферментів.
У цитоплазмі гепатоцитів з'являються дрібні краплі жиру (пилоподібне ожиріння), які зливаються у великі краплі (дрібнокрапельне ожиріння). У результаті крапля заповнює майже всю цитоплазму, відтісняючи ядро на периферію клітини (великоапельне ожиріння). Макроскопічно печінка збільшується, стає в'ялою, набуває охряно-жовтий або жовто-коричневий колір (“гусяча печінка”).
Функція печінки різко знижується. При припиненні дії фактора, що ушкоджує (наприклад, алкоголю, більше 1 місяця) можливе відновлення нормальної структури клітини. Великокапельне ожиріння веде до загибелі гепатоцитів, мезенхімальної реакції та розвитку фіброзу з результатом цирозу.
Жирова дистрофія нирок частіше розвивається у клітинах епітелію проксимальних та дистальних канальців. шляхом інфільтрації (при гіперліпідемії), рідше – декомпозиції (при наростаючій гіпоксії). Найчастіше жирова дистрофія нирок трапляється при нефротичному синдромі. Нирки при цьому збільшені, в'ялі, на розрізі з сіро-жовтим кропом. Функція нирок знижується.
Є група спадкових системних ліпідозів, що становлять хвороби накопичення (тезаурісмози). Ці захворювання пов'язані зі спадковими дефектами ферментів, які здійснюють метаболізм складних жирів. Сюди відносять: цереброзидліпідоз (хвороба Гоше), сфінгомієлінліпідоз (хвороба Німанна-Піка), гангліозидліпідоз (хвороба Тея-Сакса або рання амавротична ідіотія), генералізований гангліозидоз (хвороба Нормана-Ландінга) та ін. Центральна нервова система. Морфологічному діагнозу допомагають виявлені в тканинах характерні для того чи іншого виду ліпідозу клітини (клітини Гоше, клітини Піка)
Паренхіматозні вуглеводні дистрофії. Характеризуються порушенням обміну глікогену чи глікопротеїдів.
Порушення обміну глікогену розвивається при цукровому діабеті. При цьому захворюванні відзначається абсолютна та відносна недостатність інсуліну, внаслідок чого порушується утилізація глюкози та синтез глікогену. В результаті підвищується вміст глюкози в крові (гіперглікемія), з'являється глюкоза в сечі (глюкозурія), виснажуються запаси глікогену в печінці та м'язах. У зв'язку з гіперглікемією та глюкозурією відбувається інфільтрація глюкозою канальцевого епітелію нирок та синтез глікогену в епітелії канальців, де його в нормі не буває. Цитоплазма клітин канальців нирок стає світлою та пінистою.
Надмірне накопичення глікогену в клітинах печінки, нирок, скелетної мускулатури також виникає при спадкових хворобах накопичення (тезаурісмозі) - глікогенозах, обумовлених недостатністю різних ферментів, що регулюють обмін глікогену (хвороби Гірке, Андерсена, Помпе, Мак-Ардля та ін.).
При порушенні обміну глікопротеїдів у клітинах накопичуються муцини та мукоїди (слизоподібні речовини). Це спостерігається зазвичай при запальних процесах у слизових оболонках, наприклад, при ринітах, гастритах, бронхітах і т.д. Такий слиз може закривати просвіти бронхів, проток залоз, що веде до утворення кіст. Іноді продукуються слизоподібні колоїдні речовини у щитовидній залозі при колоїдному зобі. Закінчується процес атрофією слизових оболонок. Надмірне накопичення слизу спостерігається також при муковісцидозі (системне спадкове захворювання). При цьому клітини епітелію залоз підшлункової залози, бронхіального дерева, травного та сечового трактів, потових та слізних залоз, жовчних шляхів) продукують густий в'язкий слиз. Просвіти залоз кістозно розширюються, навколо них відбувається розростання сполучної тканини – кістозний фіброз.
СТРОМАЛЬНО-СУДИННІ (мезенхімальні) ДИСТРОФІЇ
Мезенхімальні дистрофії виявляються у стромі органів та стінках кровоносних судин, і залежно від виду порушення обміну поділяються на білкові, жирові та вуглеводні.
До мезенхімальних диспротеїнозвідносять: борошно набухання, фібриноїдне набухання, гіаліноз і амілоїдоз.
При мукоїдному набуханні в основному речовині сполучної тканини (частіше в стінках кровоносних судин, ендокарді, синовіальних оболонках) відбувається накопичення і перерозподіл глікозаміногліканів, які мають властивість притягувати воду, а так само плазмових білків, переважно глобулінів.
Колагенові волокна при цьому набухають, розволокаються, але залишаються збереженими. Глікозаміноглікани, що накопичуються, мають феномен метахромазії (здатність змінювати основний тон забарвлення), який легко дозволяє визначати вогнища мукоїдного набухання в сполучній тканині. Найбільш чітко цей феномен проявляється при забарвленні толуїдиновим синім, коли осередки борошна набухання забарвлюються не в синій, а в бузковий або червоний колір.
Ця дистрофія найчастіше розвивається при інфекційно-алергічних (гломерулонефрит), алергічних (реакції гіперчутливості негайного типу) та аутоімунних (ревматичні хвороби) захворюваннях. Зовнішній вигляд органу при цьому не змінений, і набухання мукоидное виявляється тільки мікроскопічно. При усуненні причини може бути оборотним, інакше дезорганізація сполучної тканини наростає і надалі розвивається фібриноїдне набухання.
При фібриноїдному набуханніпродовжується просочування тканин білками плазми, які накопичуються в основному речовині та колагенових волокнах, руйнуючи їх і перетворюючи на гомогенну масу, що містить фібриноїд-складна речовина, що складається з фібрину, полісахаридів, імунних комплексів (при ревматизмі), ну. Метахромазія сполучної тканини вже не виявляється, оскільки сталося руйнування глікозаміногліканів основної речовини. Фібриноїдне набухання - незворотний процес, в результаті якого можуть розвинутись фібриноїдний некроз, склероз або гіаліноз.
При гіалінозіу сполучній тканині утворюються однорідні напівпрозорі щільні білкові маси, що нагадують гіаліновий хрящ (гіалін).
Гіаліноз може бути загальним та місцевим. Також розрізняють гіаліноз судин і власне сполучної тканини.
Гіаліноз може бути результатом трьох станів: фібриноїдного набухання та некрозу, склерозу та плазматичного просочування.
Гіаліноз судинвражає дрібні артерії та артеріоли і виникає в результаті плазматичного просочування при артеріальній гіпертензії та цукровому діабеті. Внаслідок відкладення у стінці білків плазми крові судини втрачають еластичність, просвіт їх звужується, у результаті погіршується кровопостачання органу. Гіаліноз судин - системний процес і найбільш виражений у нирках, головному мозку, сітківці ока, підшлунковій залозі, шкірі. Особливо небезпечний гіаліноз артеріол головного мозку, оскільки раптові підйоми артеріального тиску (гіпертонічні кризи) можуть призвести до розриву судини та крововиливу в речовину головного мозку.
Гіаліноз власне сполучної тканинирозвивається у результаті фібриноїдного набухання чи склерозу. Як результат фібриноїдного набухання гіаліноз зустрічається при захворюваннях, пов'язаних з імунними порушеннями, наприклад, при ревматичних хворобах і зазвичай завершує прогресуючу деструкцію сполучної тканини. При ревматизмі гіаліноз виникає у стулках клапанів серця та хордах, що сприяє формуванню вад серця.
Як результат склерозугіаліноз утворюється у плеврі, листках перикарду, очеревині після перенесених запальних процесів. Гіаліноз капсули селезінки призводить до утворення “глазурної селезінки” – капсула стає товстою, просоченою білковими масами. Гіалін також може відкладатися в сполучну тканину шкіри після її ушкодження, особливо після опіків в області обличчя та шиї, що призводить до формування грубих, щільних, білих рубців (келоїдні рубці).
Амілоїдоз.
Амілоїдоз (лат. amylum – крохмаль) – стромально-судинний диспротеїноз, що супроводжується глибокими змінами білкового обміну та появою аномального фібрилярного білка – амілоїду.
У 1884 році Рокитанський назвав амілоїдоз сальною хворобою, т.к. уражені органи мають сальний вигляд. Пізніше Р Вірхов показав, що під дією йоду та сірчаної кислоти ця речовина забарвлюється у синій колір і запропонував називати його амілоїдом. Білкова природа амілоїду була встановлена в 1865 (М.Руднєв, Кюне).
Мікроскопічно при фарбуванні гематоксиліном та еозином амілоїд виглядає як еозинофільні щільні безструктурні маси. Для того, щоб відрізнити амілоїд від інших відкладень використовують гістохімічні методи, наприклад, забарвлення конго червоним, іодгрюн, генціанвіолет і метилвіолет, засновані на феномен метахромазії, так як у складі амілоїдних мас містяться глікозаміноглікани.
Виділяють такі основні види амілоїду в залежності від його хімічної будови:
АА амілоїд, який виявляється при деяких видах спадкового (сімейна середземноморська лихоманка) та при вторинному амілоїдозі. В результаті активації системи моноцитарних фагоцитів у печінці під впливом інтерлейкіну-1 відбувається стимуляція синтезу SAA, надалі відбувається його деградація з утворенням білка AA з якого на поверхні макрофагів (амілоїдобластів) йде складання фібрил амілоїду;
AL амілоїд, що виявляється при первинному амілоїдозі та при неопластичній плазмоклітинній дискразії. Спочатку відбувається синтез легких ланцюгів імуноглобулінів з яких плазматичними клітинами, макрофагами та мієломними клітинами та синтезуються фібрили амілоїду;
FAP (AF) амілоїд виявляється при деяких видах спадкового амілоїдозу (сімейна амілоїдна полінейропатія)
AS амілоїд, що виникає при сенільному амілоїдозі
Утворення амілоїду тісно пов'язане з волокнами сполучної тканини. Амілоїд може відкладатися по ходу як ретикулярних волокон (периретикулярний амілоїдоз), наприклад, у селезінці, печінці, нирках, кишечнику, так і по ходу колагенових волокон (периколагеновий амілоїдоз), наприклад у поперечно-смугастих і гладких м'язах, шкірі.
Амілоїдоз так само поділяють за переважним відкладенням амілоїду в органах на нефропатичний, кардіопатичний, нейропатичний, гепатопатичний і т.д.
Виділяють такі клініко-анатомічні форми амілоїдозу:
Ідіопатичний (первинний) амілоїдоз. Причина та механізм не відомі. При цьому амілоїд з'являється в стромі міокарда та стінках судин (кардіопатичний амілоїдоз), а також по ходу периферичних нервів (нейропатичний амілоїдоз)
Спадковий (генетичний, сімейний) амілоїдоз.Це кардіопатичний, нейропатичний, рідше – нефропатичний варіанти. Найбільш поширений у країнах Середземномор'я (Ізраїль, Ліван тощо)
Старечий амілоїдоз. Амілоїд відкладається у корі великих півкуль головного мозку, будучи центром сенільних (старечих) бляшок, а також у стінці дрібних кровоносних судин. Такі зміни у великій кількості з'являються при сенільному (старечому) недоумстві та хворобі Альцгеймера.
Придбаний (вторинний) амілоїдоз. Зустрічається найчастіше і сприймається як ускладнення різних захворювань, що супроводжуються хронічними нагноительними і деструктивними процесами. Амілоїд відкладається на тлі будь-якого захворювання: туберкульоз; хронічне гнійне запалення, наприклад, неспецифічні захворювання легень (бронхоектатична хвороба, хронічні пневмонії), остеомієліт, хронічні абсцеси, хроніосепсис, а також ревматичні хвороби (насамперед ревматоїдний артрит); та пухлини крові (мієломна хвороба). Амілоїд при цьому зазвичай відкладається в нирках, селезінці, печінці, надниркових залозах, кишечнику.
Органи збільшуються у розмірах, стають щільними, але в розрізі мають сальний вигляд.
У селезінці амілоїд відкладається спочатку в лімфатичних фолікулах у вигляді напівпрозорих зерен - сагова селезінка, потім накопичується і в червоній пульпі, селезінка при цьому збільшується, стає щільною, поверхня розрізу гладка, блискуча - сальна селезінка.
Амілоїдоз нирок має велике значення в клініці, оскільки небезпечний для життя хворого. Почавшись у пірамідах мозкової речовини нирок, амілоїдоз поступово захоплює кіркову речовину. Амілоїд відкладається у дрібних судинах, у мезангії клубочків, на базальній мембрані канальців, у стромі органу.
Нирка збільшується у розмірах, речовина її щільна, біла, із сальним блиском (“велика сальна нирка”). Виникає клінічна та морфологічна картина амілоїдного нефрозу, що призводить до хронічної ниркової недостатності.
Печінка при амілоїдозі також збільшена, щільна, виглядає "сальною".
Рідше амілоїд відкладається в надниркових залозах, зазвичай у його кірковому шарі, і в кишечнику, в області підслизового шару.
Функціональне значення визначається ступенем розвитку амілоїдозу. Виражений амілоїдоз веде до дистрофії та атрофії паренхіми та склерозу строми органів, до їх функціональної недостатності. При вираженому амілоїдозі найчастіше спостерігається хронічна ниркова, рідше – печінкова, серцева, легенева, надниркова, кишкова (синдром порушеного всмоктування) недостатність.
Дистрофія - складний патологічний процес, в основі якого лежить порушення тканинного метаболізму, що веде до структурних змін.
Трофіка – сукупність механізмів. визначальних метаболізм і структурну організацію клітини (тканини), необхідні виконання спеціалізованої функції.
Причини дистрофій:
1) розлади ауторегуляції клітини, які можуть бути спричинені гіперфункцією, токсичними речовинами, радіацією, недостатністю ферменту тощо.
2) порушення функції транспортних систем, що забезпечують метаболізм і структурну безпеку тканин, викликають гіпоксію.
3) порушення ендокринної, нервової регуляції
Морфогенез дистрофій:
1) інфільтрація
Надмірне накопичення речовини (нормальної, не аномальної) в результаті надлишкового синтезу.
Приклад: жировий гепатоз печінки, гемосидероз нирки.
2) декомпозиція (фанероз)
Розпад ультраструктур клітин та міжклітинної речовини, що веде до порушення тканинного метаболізму та накопичення продуктів порушеного обміну в тканині.
3) збочений синтез
Синтез аномальних продуктів. До них відносяться: синтез аномального білка амілоїду у клітині, синтез білка алкогольного гіаліну гепатоцитом.
4) трансформація
Утворення продуктів одного виду обміну із загальних вихідних продуктів, що йдуть на побудову БЖУ.
Класифікація дистрофії.
У класифікації дотримуються кількох принципів. Вирізняють дистрофії:
1) з переважання морфологічних зміну тканинних структурах: паренхіматозні, змішані, мезенхімальні (стромально-судинні)
2) з переважання порушень того чи іншого виду обміну. білкові, жирові, вуглеводні, мінеральні.
3) залежно від впливу генетичних факторів. набуті, спадкові.
4) по локалізації. місцеві, загальні.
Паренхіматозні дистрофії.
Прояви порушень обміну у високоспеціалізованих у функціональному відношенні клітинах.
1) Паренхіматозні білкові дистрофії (диспротеїнози)
Сутність таких дистрофій полягає у зміні фізико-хімічних та морфологічних властивостей білків клітини: вони піддаються денатурації та коагуляції або коліквації, що веде до гідратації цитоплазми. У тих випадках, коли порушаться зв'язки білків із ліпідами, виникає деструкція мембранних структур клітини.
Порушення обміну білків часто поєднується з розладами роботи Na-K-помпи: що веде до накопичення іонів Nа та набухання клітини. Такий патологічний процес називається гідропічною дистрофією.
-зерниста
Оборотна, виглядає як накопичення дрібних зерен білка в цитоплазмі. Органи збільшуються в розмірах, стають в'ялими та тьмяними.
-гіаліново-краплинна
У цитоплазмі з'являються великі гіаліноподібні білкові краплі, що зливаються між собою і клітини, що заповнюють тіло. У ряді випадків завершується фокальним коагуляційним некрозом клітини.
Часто зустрічається у нирках, рідко – у печінці та міокарді.
У нирках для дослідження накопичення крапель знаходять у нефроцитах. Накопичення часто відзначається при нефротичному синдромі, оскільки основу цієї дистрофії лежить недостатність вакуолярно-лизосомального апарату епітелію проксимального канальця, у якому нормі реабсорбуються білки. Саме тому в сечі з'являється білок (протеїнурія) та циліндри (циліндрурія).
Зовнішній вигляд не має якихось характерних рис.
У печінці при мікроскопії виявляються тільця Мелорі, що складаються з фібрил та алкогольного гіаліну. Поява таких крапель – прояв збоченої синтетичної функції гепатоциту, що зустрічається при алкогольному гепатиті, первинному біліарному цирозі. Зовнішній вигляд печінки різний.
Результат гіаліново-крапельної дистрофії несприятливий, вона веде до некрозу клітини.
-гідропічна дистрофія
Характеризується поява у клітині вакуолей, наповнених цитоплазматичною рідиною. Спостерігається частіше в епітелії шкіри та ниркових канальців, у гепатоцитах та міоцитах.
Паренхіматозні клітини збільшені в обсязі, їхня цитоплазма заповнена вакуолями, що містять прозору рідину. Потім клітина перетворюється на величезний балон (вся клітина стала великою вакуоллю) – фокальний некроз коліквації. Зовнішній вигляд тканин мало змінюється.
Велику роль у механізмі розвитку відіграє порушення проникності мембрани, що веде до закислення цитоплазми, активації гідролітичних ферментів лізосом, які розривають внутрішньомолекулярні зв'язки із приєднанням води.
Причини: у нирках – пошкодження ниркового фільтру, що веде до гіперфільтрації, у печінці – гепатити різної етіології, в епідермісі – набряк, інфекція.
Результат такої дистрофії, як правило, несприятливий – вона завершується фокальним коагуляційним некрозом.
-рогова дистрофія
Характеризується надлишковим утворенням рогової речовини в ороговіє епітелії (гіперкератоз, іхтіоз) або утворення рогової речовини там, де в нормі його не буває (патологічне зроговіння на слизових оболонках). Причини різноманітні: порушення розвитку шкіри, хронічне запалення, авітаміноз тощо.
Вихід: іноді при усуненні причини відбувається відновлення тканини, однак у запущених випадках настає загибель клітин.
- Спадкові порушення обміну амінокислот
Так звані хвороби накопичення, в основі яких лежить порушення внутрішньоклітинного метаболізму ряду амінокислот у результаті спадкової недостатності метаболізуючих ферментів.
а) цистиноз. Науці ще відомо, недостатність якого ферменту призводить до цього захворювання. АК накопичується в печінці, нирках, селезінці, очах, кістковому мозку, шкірі.
Б) тирозиноз. Виникає при дефіциті тирозинамінотрансферази. Накопичується у печінці, нирках, кістках.
В) фенілпіровіноградна олігофренія. Виникає при дефіциті фенілаланін-4-гідроксилази і накопичується в нервовій системі, м'язах та крові.
2) Паренхіматозні жирові дистрофії (ліпідози)
Порушення обміну цитоплазматичних ліпідів можуть виявлятися у збільшенні їхнього вмісту в клітинах, де вони виявляються і в нормі. у появі ліпідів там, де вони зазвичай не зустрічаються, та у освіті жирів незвичайного хімічного складу.
-порушення обміну ліпідів
У печінці жирова дистрофія проявляється різким збільшенням вмісту жирів у гепатоцитах та зміною їх складу. У клітинах печінки спочатку з'являються гранули ліпідів (пилоподібне ожиріння), потім дрібні краплі (дрібнокрапельне ожиріння), які потім зливаються у великі краплі (великокропельне) або одну жирову вакуоль. Печінка збільшена, в'яла та охряно-жовтого кольору. Серед механізмів жирової дистрофії печінки розрізняють надмірне надходження в гепатоцити жирних кислот або підвищений їх синтез цими клітинами, вплив токсичних речовин, що блокують окислення жирних кислот і синтез ліпопротеїдів у гепатоцитах, недостатнє надходження в печінкові клітини амінокислот, необхідних для синтезу. Отже, ЗДП виникає в результаті: ліпопротеїдемії (алкоголізм, цукровий діабет, загальне ожиріння), гепатотропних інтоксикаціях (етанол, хлороформ), порушення харчування.
Жирова дистрофія міокарда виникає внаслідок гіпоксії та інтоксикації. Механізм розвитку пов'язаний із зниженням окислення жирних кислот через деструкцію мітохондрій під дією гіпоксії чи токсину. При макроскопічному дослідженні розміри серця збільшені, серцевий м'яз глиняно-жовтого кольору. Міокард схожий на шкіру тигра – біло-жовта смугастість. Ліпіди визначаються у вигляді дрібних крапель.
Причини жирової дистрофії різноманітні. Вони можуть бути пов'язані з кисневим голодуванням (тому часто зустрічається при захворюваннях ССС), інфекціями та інтоксикаціями, авітамінозами та одностороннім харчуванням.
Результат жирової дистрофії залежить від її ступеня. Якщо не супроводжується грубою статтю клітинних структур, то вона оборотна.
-спадкові ферментопатії
Виникають унаслідок спадкового дефіциту ферментів, що у метаболізмі ліпідів.
А) хвороба Гоше при дефіциті глюкоцереброзідази. Ліпід накопичується в печінці, селезінці, кістковому мозку.
Б) хвороба Німанна-Піка при дефіциті сфінгомієлінази. Накопичення в печінці, селезінці, кістковому мозку.
В) хвороба Саксу при дефіциті кислої галактозидази.
Г) хвороба Нормана-Ландінга при дефіциті бета-галактозидази.
3) Паренхіматозні вуглеводні дистрофії
-вуглеводні дистрофії, пов'язані з порушенням обміну глікогену
При цукровому діабеті відбувається недостатнє використання глюкози тканинами, збільшення її вмісту у крові та виведення із сечею. Тканинні запаси глікогену різко зменшуються. У печінці порушується синтез глікогену, що веде до її інфільтрації жирами та жирової дистрофії печінки.
У нирках при цукровому діабеті відбуваються такі зміни: - глікогенна інфільтрація епітелію канальців.
-спадкові глікогенози
а) 1 типу – хвороба Гірке – дефіцит глюкозо-6-фосфатази
б) 2 типи – хвороба Помпе – дефіцит кислої альфа-1,4-глюкозидази
в) 3 типи – хвороба Форбса – дефіцит аміло-1,6-глюкозидази
г) 4 типи – хвороба Андресона – дефіцит аміло-(1,4-1,6)-трансглюкозидази
д) 5 типу – хвороба Мак-Ардля – дефіцит міофосфорилази
е) 6 типу – хвороба Герса – дефіцит фосфорилази печінки
За хвороб 1,2,5,6 типів структура глікогену не порушена.
-вуглеводні дистрофії, пов'язані з порушеннями обміну глікопротеїдів
У клітинах або міжклітинній речовині відбувається накопичення муцинів і мукоїдів, званих також слизовими або слизовими речовинами.
Багато клітин, що секретують, гинуть і десквамуються, вивідні протоки залоз обтуруються слизом, що веде до розвитку кіст.
Причини різноманітні, але найчастіше запалення слизових оболонок внаслідок дії різних патогенних подразників.
Більше інформації
Зерниста дистрофія нирки (мікрокартина) Зерниста дистрофія (мутне набухання) нирки. Мікрокартина. Забарвлення Г-Е. (по Ст А. Салімову) А (ув. 240): 1. ядра клітин епітелію ниркових канальців; 2. судинний клубочок; 3. звивисті канальці; 4. скупчення білка (білкові циліндри) у просвіті звивистого канальця.
 Зерниста дистрофія нирки (мікрокартина) Зерниста дистрофія (мутне набухання) нирки. Мікрокартина. Забарвлення Г-Е. (по В. А. Салімову) Б (ув. 960): 1. епітелій звивистих канальців (відзначається дрібна, пилоподібна зернистість цитоплазми, клітини набряклі, відсутні ядра, межі між клітинами не проглядаються); 2. білковий циліндр; 3. нирковий (судинний) клубочок
Зерниста дистрофія нирки (мікрокартина) Зерниста дистрофія (мутне набухання) нирки. Мікрокартина. Забарвлення Г-Е. (по В. А. Салімову) Б (ув. 960): 1. епітелій звивистих канальців (відзначається дрібна, пилоподібна зернистість цитоплазми, клітини набряклі, відсутні ядра, межі між клітинами не проглядаються); 2. білковий циліндр; 3. нирковий (судинний) клубочок
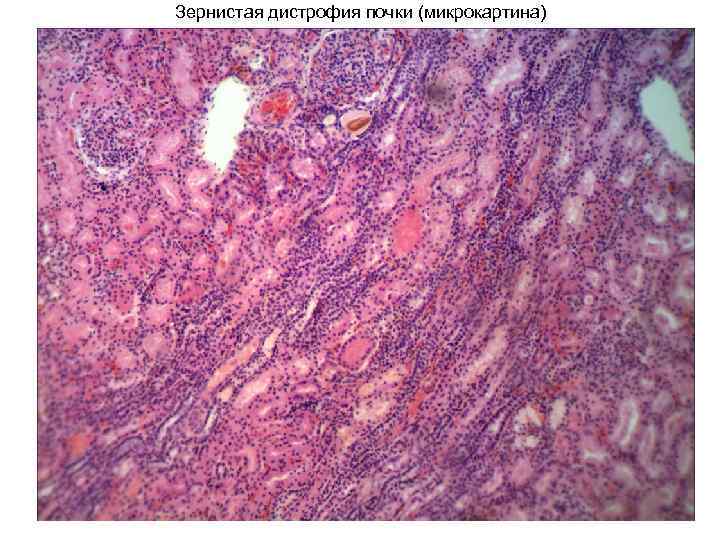

 Зерниста дистрофія нирки (макрокартина) Зерниста дистрофія (мутне набухання) нирки. Макрокартіна. (за В. А. Салімову) 1. кірковий шар; 2. згладжена межа між кірковим та мозковим шарами нирки; 3. мозковий шар нирки; 4. нирка з поверхні; 5. крововиливи в нирку
Зерниста дистрофія нирки (макрокартина) Зерниста дистрофія (мутне набухання) нирки. Макрокартіна. (за В. А. Салімову) 1. кірковий шар; 2. згладжена межа між кірковим та мозковим шарами нирки; 3. мозковий шар нирки; 4. нирка з поверхні; 5. крововиливи в нирку
 Зерниста дистрофія нирки (мікрокартина) 1. Нирковий канал без видимих змін; 2. Ниркові канальці в стані зернистої дистрофії: А) скупчення пилоподібної зернистості білкової природи в клітинах епітелію ниркових канальців, Б) химерно змінений просвіт ниркового канальця
Зерниста дистрофія нирки (мікрокартина) 1. Нирковий канал без видимих змін; 2. Ниркові канальці в стані зернистої дистрофії: А) скупчення пилоподібної зернистості білкової природи в клітинах епітелію ниркових канальців, Б) химерно змінений просвіт ниркового канальця


 Зерниста дистрофія печінки 1. Гепатоцити у стані зернистої дистрофії та некробіозу; 2. Цитоплазма гепатоцитів, збільшена в обсязі, неоднорідна, з дрібною пилоподібною зернистістю та просвітленнями; 3. Гепатоцити, що втратили ядро (у стані некробіозу)
Зерниста дистрофія печінки 1. Гепатоцити у стані зернистої дистрофії та некробіозу; 2. Цитоплазма гепатоцитів, збільшена в обсязі, неоднорідна, з дрібною пилоподібною зернистістю та просвітленнями; 3. Гепатоцити, що втратили ядро (у стані некробіозу)




 Вакуольна (гідропічна) дистрофія нирки. Мікрокартина. Забарвлення Г-Е 1. Наявність балонів та вакуолей у цитоплазмі клітин ниркового епітелію; 2. Розпад ядер та цитоплазми в результаті здавлювання їх вакуолями (формування «персневидних клітин»)
Вакуольна (гідропічна) дистрофія нирки. Мікрокартина. Забарвлення Г-Е 1. Наявність балонів та вакуолей у цитоплазмі клітин ниркового епітелію; 2. Розпад ядер та цитоплазми в результаті здавлювання їх вакуолями (формування «персневидних клітин»)


 Гіаліново-краплинна дистрофія нирки. Мікрокартина Гіаліново-краплинна дистрофія нирки. Мікрокартина. Забарвлення Г-Е. (за В. В. Сєрову, Н. Є. Яригіна, В. С. Паукова) а. цитоплазма клітин епітелію ниркових канальців заповнена великими краплями білкової природи
Гіаліново-краплинна дистрофія нирки. Мікрокартина Гіаліново-краплинна дистрофія нирки. Мікрокартина. Забарвлення Г-Е. (за В. В. Сєрову, Н. Є. Яригіна, В. С. Паукова) а. цитоплазма клітин епітелію ниркових канальців заповнена великими краплями білкової природи
 Гіаліново-краплинна дистрофія нирки. Мікрокартина 1. Скупчення гіалінових крапель у цитоплазмі клітин ниркового епітелію канальців (це скупчення викликає атрофію та розпад цитоплазми та ядра); 2. вихід гіалінових крапель у просвіт ниркових канальців
Гіаліново-краплинна дистрофія нирки. Мікрокартина 1. Скупчення гіалінових крапель у цитоплазмі клітин ниркового епітелію канальців (це скупчення викликає атрофію та розпад цитоплазми та ядра); 2. вихід гіалінових крапель у просвіт ниркових канальців



 Гіаліноз судин матки 1. Кровоносна судина в стані гіалінозу; 2. Різко потовщена стінка судини внаслідок відкладення у ній гіаліну; 3. «Тканинна муфта»; 4. Різко звужений щілинний просвіт судини; 5. Маткові залози, що збереглися.
Гіаліноз судин матки 1. Кровоносна судина в стані гіалінозу; 2. Різко потовщена стінка судини внаслідок відкладення у ній гіаліну; 3. «Тканинна муфта»; 4. Різко звужений щілинний просвіт судини; 5. Маткові залози, що збереглися.


 Гіаліноз капсули і строми селезінки 1. Відкладення гіаліну в капсулі селезінки (капсула набуває гомогенного вигляду і різко збільшується в обсязі); 2. Поздовжній та поперечний зріз трабекул селезінки, які просякнуті гіаліном; 3. Паренхіма селезінки без змін.
Гіаліноз капсули і строми селезінки 1. Відкладення гіаліну в капсулі селезінки (капсула набуває гомогенного вигляду і різко збільшується в обсязі); 2. Поздовжній та поперечний зріз трабекул селезінки, які просякнуті гіаліном; 3. Паренхіма селезінки без змін.
 Амілоїдоз селезінки (сагова форма). Мікрокартина. Забарвлення Г-Е. Амілоїдоз селезінки (сагова форма). Мікрокартина. Забарвлення Г-Е. (Х 240). (за Ст А. Салімову) 1. фолікули білої пульпи; 2. атрофована червона пульпа; 3. ядра збережених клітин; 4. відкладення амілоїду у фолікулах білої пульпи
Амілоїдоз селезінки (сагова форма). Мікрокартина. Забарвлення Г-Е. Амілоїдоз селезінки (сагова форма). Мікрокартина. Забарвлення Г-Е. (Х 240). (за Ст А. Салімову) 1. фолікули білої пульпи; 2. атрофована червона пульпа; 3. ядра збережених клітин; 4. відкладення амілоїду у фолікулах білої пульпи

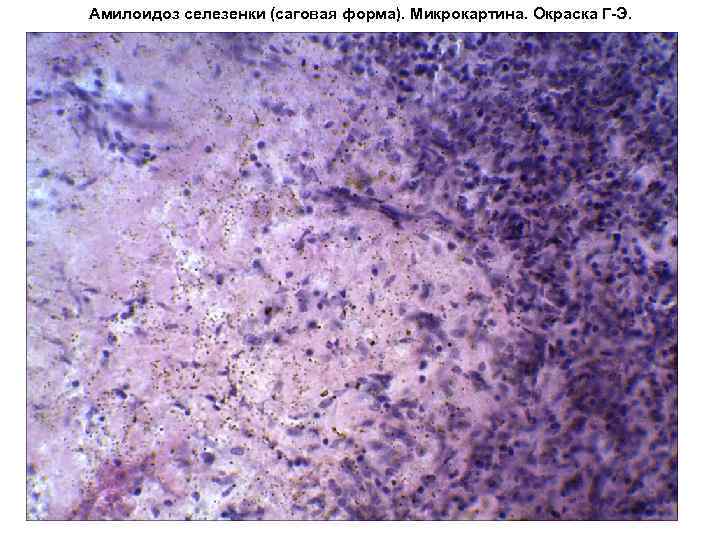


 Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е. Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е. (Х 480). (за В. А. Салімову) 1. дифузне відкладення амілоїду; 2. атрофована пульпа.
Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е. Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е. (Х 480). (за В. А. Салімову) 1. дифузне відкладення амілоїду; 2. атрофована пульпа.
 Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е.
Амілоїдоз селезінки (сальна форма). Мікрокартина. Забарвлення Г-Е.
Залежно від того, в яких структурах переважно локалізуються дистрофічні зміни, їх ділять на паренхіматозні, стромально-судинні (мезенхімальні), змішані.
Залежно від переважання порушень тієї чи іншої видів обміну виділяють білкові, жирові, вуглеводні, мінеральні дистрофії, змішані.
Також розрізняють набуті та спадкові дистрофії, і залежно від поширеності – загальні та місцеві.
Паренхіматозні дистрофії
При цих дистрофіях речовини накопичуються в клітинах паренхіми різних органів, таких як міокардіоцити, гепатоцити, нейрони, клітини ниркових канальців, надниркових залоз. За видом порушеного обміну ці дистрофії ділять на білкові (диспротеїнози), жирові (ліпідози) та вуглеводні.
Паренхіматозні білкові дистрофії (диспротеїнози) характеризуються порушенням обміну цитоплазматичних білків, що у вільному чи пов'язаному стані. До них відносяться зерниста, гідропічна, гіаліново-крапельна та рогова дистрофії.
Зерниста дистрофія.
Причинами зернистої дистрофії можуть бути розлади кровообігу, інфекції, інтоксикації та інші фактори, що ведуть до зниження інтенсивності окисно-відновних процесів клітини. Органи при цьому процесі збільшені, в'ялі, на розрізі тьмяні, каламутні. Це дає підставу називати зернисту дистрофію каламутним (тьмяним) набуханням органів.
Мікроскопічно клітини збільшуються, цитоплазма їхня каламутна, багата на білкові гранули. Електронно-мікроскопічно при зернистій дистрофії відзначається збільшення кількості (гіперплазія) та набухання органел клітини, які світлооптично виглядають як білкові гранули. Така гіперплазія ультраструктур клітин в даний час розглядається як виражена функціональна напруга органів на різні дії.
Вихід зернистої дистрофії різний. У більшості випадків вона оборотна, але якщо причини, що її викликали, не усунуті, можливий її перехід у гіаліново-краплинну, гідропічну або жирову дистрофію.
 (1оцінок, у середньому: 5,00із 5)
(1оцінок, у середньому: 5,00із 5)