Види хромосомних мутацій. Хромосомні мутації: приклади. Види хромосомних мутацій. загальні положення
Геном людини: Енциклопедія, написана чотирма літерами Тарантул В'ячеслав Залманович
Хромосома 5
Хромосома 5
Більшість генів цієї хромосоми сконцентровано у двох областях довгого плеча та одному районі короткого ближче до його кінця. Є два райони, розташовані навколо центроміри, збагачені сніпсами. C генами хромосоми 5 пов'язаний ряд важких захворювань: мегалопластична анемія, колоректальний рак, капілярна гемангіома, дистрофія рогівки, аутосомно-домінантна глухота, синдром Гарднера, хвороба Гіршспрунга, кетоацитоз, гостра промієлома ін.
З книги Геном людини: Енциклопедія, написана чотирма літерами автора Тарантул В'ячеслав ЗалмановичХромосома 2 Це друга за розмірами хромосома. Найбільша щільність сніпсів є в районі центроміру, а повтори тут практично відсутні. На одиницю довжини у ній міститься помітно менше генів, ніж у хромосомі 1 та інших хромосом. Тим не менш, число
З книги автораЦе ще одна досить велика хромосома. На відміну від хромосоми 2 в неї в області центроміру міститься мало як сніпсів, так і повторів. Найбільша кількість сніпсів розташована ближче до кінців цієї хромосоми, а найбільше генів - на короткому плечі.
З книги автораХромосома 4 Гени, повтори та сніпси розподілені в хромосомі 4 досить рівномірно (за винятком району центроміру, де всі вони представлені малою кількістю). Підраховано, що загальна кількість генів тут менша, ніж у середньому на одиницю довжини геному. Серед захворювань,
З книги автораХромосома 5 Більшість генів цієї хромосоми сконцентровано у двох областях довгого плеча та одному районі короткого ближче до його кінця. Є два райони, розташовані навколо центроміри, збагачені сніпсами. C генами хромосоми 5 пов'язаний ряд важких захворювань:
З книги автораХромосома 6 Щільність генів і сніпсів найбільша в декількох районах на короткому плечі цієї хромосоми, а ось повтори розподілені вздовж хромосоми досить рівномірно (їх мало тільки в області центроміри). C генами хромосоми 6 пов'язаний ряд патологій людини: діабет,
З книги автораХромосома 7 Щільність сніпсів найбільша у прицентромірній ділянці довгого плеча цієї хромосоми. А ось гени розташовані досить рівномірно вздовж хромосоми, за винятком однієї ділянки в середині довгого плеча, де міститься їх найбільша кількість. Серед
З книги автораХромосома 8 Більшість сніпсів у цій хромосомі сконцентровано на кінці короткого плеча, а на кінці довгого плеча є область сильно збагачена генами. Число генів, асоційованих із захворюваннями, у хромосомі 8 відносно невелике. Серед них є гени,
З книги автораХромосома 9 Тут і сніпси, і повтори, і гени розподілені дуже нерівномірно вздовж хромосоми. Крім того, хромосома 9 збагачена сніпс у порівнянні з іншими хромосомами (при розрахунку їх числа на одиницю довжини). При цьому найбільша їх кількість сконцентрована в
З книги автораХромосома 10 Ця хромосома є середньою за кількістю генів, що містяться в ній, повторюваних ділянок і сніпсів на одиницю довжини, але розподіл їх по хромосомі далеко не рівномірний: кілька ділянок на довгому плечі сильно збагачені генами і сніпсами. Серед
З книги автораХромосома 11 На кінці короткого плеча та в прицентромірному районі довгого плеча цієї хромосоми має місце концентрація генів. Зміст сніпсів підвищено лише в районі кінця короткого плеча, а вздовж хромосоми він є однаковим. Від загальної кількості генів цієї
З книги автораХромосома 12 Ця хромосома є середньою за більшістю параметрів. Гени розподілені у ній дуже нерівномірно. З ними асоційований ряд захворювань: адренолейкодистрофія, амілоїдозіс, злоякісна неходжкінська лімфома, рак прямої кишки, емфізема, енурез,
З книги автораХромосома 13 Коротке плече цієї хромосоми поки що погано секвеновано. Є концентрація сніпсів у районі центроміру на довгому плечі. Хромосома 13 щодо інших хромосом збіднена генами (на 1 млн. букв у ній у середньому припадає лише близько 5 генів). Найбільше їх
З книги автораХромосома 20 стала третьою за часом повністю секвенованою хромосомою людини. За розміром ця хромосома становить близько двох відсотків генетичного коду геному людини. Гени, повтори та сніпси розподілені вздовж хромосоми дуже нерівномірно.
З книги автораХромосома 21 Ця хромосома є найменшою за розмірами та інформаційною ємністю (на її частку припадає не більше 1,5% від усього геному людини). Але секвенована вона була лише за хромосомою 22. Число генів у хромосомі 21 відносно невелике. При розмірі близько
З книги автораХромосома 22 ДНК цієї хромосоми була секвенована першою (грудень 1999), тому вона і описана повніше. У хромосомі 22 залишилися нерозшифрованими лише кілька ділянок (менше 3% довжини ДНК). Вона містить близько 500 генів та 134 псевдогени. Всі ці генні послідовності
З книги автораХромосома X Це жіноча статева хромосома. Наявність двох хромосом X визначає жіноча стать. Пара для хромосоми X у чоловіків - омертвіла та коротка Y-хромосома. У жінок на одній із двох хромосом X відбувається інактивація всіх генів, які мають пари на хромосомі Y.
Хромосомні мутації (інакше їх називають абераціями, перебудовами) - це непередбачувані зміни у структурі хромосом. Найчастіше вони викликаються проблемами, що виникають у процесі поділу клітини. Вплив факторів середовища, що ініціюють, - це ще одна можлива причина хромосомних мутацій. Давайте розберемося, якими можуть бути прояви такого роду змін у структурі хромосом і які наслідки вони несуть для клітини і всього організму.
Мутації. загальні положення
У біології мутація окреслюється стійке зміна структури генетичного матеріалу. Що означає «стійке»? Воно передається у спадок нащадкам організму, що має мутантну ДНК. Відбувається це в такий спосіб. Одна клітина одержує неправильну ДНК. Вона ділиться, а дві дочірні копіюють її будову повністю, тобто вони також містять змінений генетичний матеріал. Далі таких клітин стає все більше, і якщо організм переходить до розмноження, його нащадки отримують подібний мутантний генотип.
Мутації зазвичай не проходять безвісти. Деякі з них змінюють організм настільки, що результатом цих змін стає смерть. Частина з них змушує організм функціонувати по-новому, знижуючи його здатність до адаптації та призводячи до серйозних патологій. І дуже мала кількість мутацій приносить організму користь, підвищуючи цим його здатність адаптуватися до умов навколишнього середовища.
Виділяють мутації генні, хромосомні та геномні. Така класифікація ґрунтується на відмінностях, що відбуваються у різних структурах генетичного матеріалу. Хромосомні мутації, таким чином, зачіпають будову хромосом, генні - послідовність нуклеотидів у генах, а геномні вносять зміни до геному всього організму, додаючи або забираючи цілий набір хромосом.
Поговоримо про хромосомні мутації докладніше.
Які можуть бути хромосомні перебудови?
Залежно від того, як локалізовані зміни, що відбуваються, розрізняють такі типи хромосомних мутацій.
- Внутрішньохромосомні – перетворення генетичного матеріалу в межах однієї хромосоми.
- Міжхромосомні – перебудови, внаслідок яких дві негомологічні хромосоми обмінюються своїми ділянками. Негомологічні хромосоми містять різні гени і не зустрічаються у процесі мейозу.
Кожному з цих типів аберацій відповідають деякі види хромосомних мутацій.

Делеції
Делеція - це відділення чи випадання будь-якої ділянки хромосоми. Нескладно здогадатися, що цей тип мутації відноситься до внутрішньохромосомних.
Якщо відділяється крайня ділянка хромосоми, то делеція називається кінцевою. Якщо ж відбувається випадання генетичного матеріалу ближче до центру хромосоми, така делеція називається інтерстиціальною.
Цей тип мутацій може впливати на життєздатність організму. Наприклад, випадання ділянки хромосоми, що кодує певний ген, забезпечує людині несприйнятливість до вірусу імунодефіциту. Ця адаптаційна мутація виникла приблизно 2000 років тому і деяким людям, які захворіли на СНІД, вдалося вижити тільки завдяки тому, що їм пощастило мати хромосоми зі зміненою структурою.
Дуплікації
Ще один вид внутрішньохромосомних мутацій – дуплікації. Це копіювання ділянки хромосоми, яке відбувається внаслідок помилки при так званому перехресті, або кросинговері в процесі поділу клітини.
Скопійована таким чином ділянка може зберігати своє положення, повертатися на 180 °, або навіть повторюватися кілька разів, і тоді така мутація називається ампліфікацією.
У рослин кількість генетичного матеріалу може збільшуватись саме шляхом багаторазових дуплікацій. У такому разі зазвичай змінюються здібності цілого виду до адаптації, а це означає, що такі мутації мають велике еволюційне значення.
Інверсії
Також відносяться до внутрішньохромосомних мутацій. Інверсія – це поворот певної ділянки хромосоми на 180°.

Перевернута в результаті інверсії частина хромосоми може перебувати по один бік від центроміру (парацентрична інверсія) або по її сторони (перицентрична). Центромер - це так звана область первинної перетяжки хромосоми.
Зазвичай інверсії не впливають на зовнішні ознаки організму та не призводять до патологій. Існує, проте, припущення, що з жінок з інверсією певної ділянки дев'ятої хромосоми ймовірність викидня при вагітності зростає на 30 %.
Транслокації
Транслокація – це переміщення ділянки однієї хромосоми на іншу. Ці мутації належать до типу міжхромосомних. Виділяють два види транслокацій.
- Реципрокні – це обмін двох хромосом певними ділянками.
- Робертсонівські - злиття двох хромосом з коротким плечем (акроцентричним). У процесі робертсонівської транслокації короткі ділянки обох хромосом втрачаються.
Реципрокні транслокації призводять у людей до проблем із дітонародженням. Іноді такі мутації стають причиною невиношування вагітності або ведуть до появи дітей з вродженими патологіями розвитку.
Робертсонівські транслокації досить часто трапляються у людини. Зокрема, якщо транслокація відбувається за участю хромосоми 21, у плода розвивається синдром Дауна, одна з найчастіше зареєстрованих уроджених патологій.
Ізохромосоми
Ізохромосоми - це хромосоми, що втратили одне плече, але замінили його на точну копію іншого свого плеча. Тобто, по суті, такий процес можна вважати делецією та інверсією в одному флаконі. У дуже поодиноких випадках такі хромосоми мають дві центроміри.
Ізохромосоми присутні в генотипі жінок, які страждають на синдром Шерешевського - Тернера.
Усі описані вище види хромосомних мутацій притаманні різним живим організмам, зокрема й людині. Які ж вони проявляються?
Хромосомні мутації. Приклади
Мутації можуть відбуватися в статевих хромосомах та в аутосомах (всіх інших парних хромосомах клітини). Якщо мутагенез торкається статевих хромосом, наслідки для організму, як правило, виявляються важкими. Виникають вроджені патології, які торкаються розумового розвитку індивіда і зазвичай виражаються у змінах фенотипу. Тобто зовні мутантні організми від нормальних.
Геномні та хромосомні мутації частіше виникають у рослин. Проте трапляються вони і в тварин, і в людини. Хромосомні мутації, приклади яких ми розглянемо нижче, виявляється у виникненні важких спадкових патологій. Це синдром Вольфа-Хіршхорна, синдром котячого крику, хвороба часткової трисомії по короткому плечу хромосоми 9, а також деякі інші.
Синдром «котячого крику»
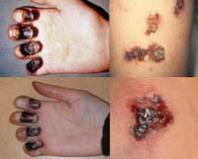
Це захворювання було відкрито 1963 року. Виникає воно через часткову моносомію по короткому плечу хромосоми 5, обумовленої делецією. Один із 45 000 дітей народжується із цим синдромом.

Чому це захворювання отримало таку назву? Діти, які страждають на цю хворобу, мають характерний плач, який нагадує котяче нявкання.
При делеції короткого плеча п'ятої хромосоми можуть втрачатися різні ділянки. Клінічні прояви захворювання безпосередньо залежить від того, які гени були втрачені під час цієї мутації.
Будова гортані змінюється у всіх хворих, а отже «котячий крик» характерний для всіх без винятку. Більшість страждають цим синдромом відзначається зміна будови черепа: зменшення мозкового відділу, луноподібна форма обличчя. Вушні раковини при синдромі "котячого крику" зазвичай розташовані низько. Іноді у хворих спостерігаються вроджені патології серця чи інших органів. Характерною ознакою стає розумова відсталість.
Зазвичай хворі з цим синдромом помирають у ранньому дитинстві, лише 10% їх доживає до десятирічного віку. Проте зафіксовано й випадки довгожительства при синдромі "котячого крику" – до 50 років.
Синдром Вольфа-Хіршхорна
Цей синдром зустрічається значно рідше – 1 випадок на 100 000 народжень. Зумовлений він делецією одного із сегментів короткого плеча четвертої хромосоми.

Прояви цього захворювання різноманітні: затримка розвитку фізичної та психічної сфери, мікроцефалія, характерна клювоподібна форма носа, косоокість, ущелини піднебіння або верхньої губи, маленький рот, вади внутрішніх органів.
Як і багато інших хромосомних мутацій людини, хвороба Вольфа-Хіршхорна відноситься до категорії напівлетальних. Це означає, що життєздатність організму за такої хвороби суттєво знижена. Діти з діагностованим синдромом Вольфа-Хіршхорна зазвичай не доживають до 1 року, проте зафіксовано один випадок, коли хворий прожив 26 років.
Синдром часткової трисомії за коротким плечем хромосоми 9
Виникає це захворювання через незбалансовані дуплікації в дев'ятій хромосомі, внаслідок чого генетичного матеріалу в цій хромосомі стає більше. Загалом відомо понад 200 випадків таких мутацій у людини.
Клінічна картина описується затримкою фізичного розвитку, легкою розумовою відсталістю, характерним виразом обличчя. Пороки серця виявляються у четвертій частині всіх хворих.
При синдромі часткової трисомії короткого плеча хромосоми 9 прогноз все ж таки відносно сприятливий: більшість хворих доживають до літнього віку.

Інші синдроми
Іноді навіть у дуже маленьких ділянках ДНК відбуваються хромосомні мутації. Хвороби в таких випадках зазвичай обумовлені дуплікаціями або делеціями, і їх називають мікродуплікаційними або мікроделеційними.
Найпоширенішим таким синдромом вважається хвороба Прадера-Віллі. Виникає вона через мікроделецію ділянки хромосоми 15. Що цікаво, ця хромосома має бути обов'язково отримана організмом від батька. Через війну мікроделеції порушеними виявляються 12 генів. У хворих із цим синдромом відзначаються розумова відсталість, ожиріння, а також у них зазвичай маленькі стопи та кисті рук.
Ще одним прикладом таких хромосомних хвороб може бути синдром Сотоса. Відбувається мікроделеція на ділянці довгого плеча хромосоми 5. Клінічна картина цього спадкового захворювання характеризується швидким зростанням, збільшенням розмірів кистей рук і стоп, наявністю опуклого чола, деякою затримкою психічного розвитку. Частота народження цього синдрому не встановлена.
Хромосомні мутації, точніше, мікроделеції на ділянках 13 та 15 хромосом, викликають відповідно пухлину Вільмса та ретинбластому. Пухлина Вільмса – це рак нирок, який виникає переважно у дітей. Ретинобластома – це злоякісна пухлина сітківки, яка також трапляється у дітей. Ці захворювання лікуються, якщо їх діагностика проведена на ранніх стадіях. У деяких випадках лікарі вдаються до оєративного втручання.

Сучасна медицина позбавляє багатьох хвороб, але вилікувати або хоча б запобігти хромосомним мутаціям поки не можна. Їх можна лише виявити на початку внутрішньоутробного розвитку плода. Проте генна інженерія не стоїть дома. Можливо, незабаром спосіб запобігання хворобам, викликаним хромосомними мутаціями, буде знайдено.
Настав час для холодного душу. Дорогий читачу, я, автор цієї книги, ввів вас в оману. Занадто часто я використовував слово «просто» і бурмотів про дивовижну простоту генетики щось на кшталт «ген - це лише пропис у «книзі рецептів» білків, написаної на диво простою мовою», пишаючись вдалою метафорою. Такий простий ген на хромосомі 3 у разі поломки викликає алкаптонурію, а інший простий ген на хромосомі 4, якщо він надто довгий - хорею Хантінгтона. Якщо в людини є мутація, вона занедужує, якщо її немає - людина здорова. Жодних дискусій, статистики та інших дурниць. І життя людини здалося нудним і накресленим. Вона, як горошини, - або гладка, або зморшкувата.
Насправді світ влаштований негаразд. Він сповнений півтонів, нюансів, специфікаторів та залежностей. Мендельська генетика так само непридатна для розуміння всієї складності та різноманіття спадковості, як евклідова геометрія для опису різноманіття форм живого дерева. За рідкісними винятками важких генетичних захворювань, якими, дякувати Богу, більшість із нас не страждає, вплив генів на наше життя вплітається тонкими волокнами в різноманіття інших факторів. Ми не ділимося на велетнів та карликів, як мендельські рослини гороху, більшість із нас – десь посередині. Ми не ділимося, як горошини, на зморшкуватих та гладких. Зморшки є у всіх, але виявляються по-різному. І в цьому немає
нічого дивного. Як вода, що складається з молекул, є не просто жменею маленьких більярдних кульок, так і людина – це не просто сума генів. Здоровий глузд підказує нам, що вплив генів далеко не так передбачувано, як розв'язання математичних рівнянь. Цікаво спостерігати, як на вашому обличчі поєднуються риси батька та матері. Але картина виходить зовсім не та, як у випадку з вашим братом чи сестрою. Кожна дитина в сім'ї все одно буде унікальною.
Ласкаво просимо у світ плейотропності та плюралізму! Ваш зовнішній вигляд визначався не тільки генами, відповідальними за цю ознаку, але й роботою всіх інших генів, крім того - багатьма негенетичними факторами, включаючи моду, ваш смак і прийняті вами вирази. Хромосома 5 - зручний об'єкт для ворожіння на кавовій гущі, щоб подивитися, як із різноманіття генів складається розмита, але багата на форми і напівтони картина спадковості. Але не стрімголов кидатися в цей світ напівтіней. Давайте рухатись крок за кроком. Я продовжу розповідь про захворювання, але цього разу йтиметься не про генетичну хворобу, та й не про хворобу зовсім, а про схильність до неї. Хромосома 5 є домівкою цілого сімейства генів, які розглядаються як головні кандидати на номінацію «генів астми». Але все, що пов'язано з ними, оповите мантією плейотропності -спеціальний термін для опису різноманітних проявів спадковості, пов'язаних із роботою численних генів. Астма – типовий приклад плейотропного захворювання. Вченим поки що не вдалося схопити за руку головний ген астми, хоч як вони намагалися.
Це захворювання у різній формі притаманне всім людям. Практично кожен з нас страждає на алергію на який-небудь подразник, якщо не з народження, то в певний період життя. Існує безліч суперечливих теорій про природу алергії. Ви можете приєднатися до будь-якої з воюючих партій. Ті, хто бореться за чистоту, звинувачують у всьому забруднення довкілля. Інші вважають, що загроза астми причаїлася в килимах, меблях та будівельних матеріалах. Хтось бачить причину астми у стресах та перевантаженнях на роботі чи у школі. Ті, хто не люблять мити руки, звинувачують у всьому нав'язливу гігієну. Іншими словами, астма – це відображення всієї складності нашого світу.
Астма – це вершина айсберга, званого атопією, -спадкової схильності до різноманітних алергій. Не дивно, що більшість астматиків ще мають алергію на продукти чи речовини. Астма, екзема, алергія та анафілаксія- це всі прояви одного синдрому, пов'язаного з роботою певних клітин організму, що активуються одними і тими ж молекулами імуноглобуліну-Е. Одна людина з десяти має клінічні прояви алергії – від легких нападів сінної лихоманки до анафілактичного шоку, який може розвинутись за лічені секунди від укусу бджоли чи горіха арахісу та призвести до смерті. Який би чинник не був причиною дедалі більшого числа астматиків, цей фактор впливає на частоту і гостроту проявів всіх інших атопійних захворювань. Відомо, що якщо у дитини була алергія, яку вона переросла, то у неї значно знижується шанс захворіти на астму у дорослому віці.
Слід зробити ще одне зауваження щодо причин астми та тверджень про стрімке зростання числа астматиків. В одних публікаціях можна прочитати, що кількість астматиків за останні 10 років зросла на 6%, а кількість людей, які страждають на алергію на арахіс, - на 7% за цей же час, причому смертність від астми вселяє побоювання. Усього кількома місяцями пізніше інші дослідники пишуть так само впевнено, що за їхніми даними приріст хворих на астму - це ілюзія. Просто люди стали більше приділяти уваги астмі, частіше звертатися до лікаря в тих випадках, в яких раніше ніколи не звернулися б і просто вважали, що застудилися. У 1870 Арман Труссо (Armand Trousseau) присвятив астмі главу своєї книги Clinique Medicate(Клінічна медицина). Він описав випадок астми у двох братів-близнюків, яких ця хвороба приковувала до ліжка у Марселі та інших містах, але повністю пройшла у Тулоні. Труссо знайшов це дуже дивним. Втім, те, що він виділив цей випадок, не свідчить про рідкість хвороби на той час. Хоча й не можна виключати, що кількість хворих на астму та алергії дійсно зростає і винна в цьому забруднення навколишнього середовища.
Але про яке забруднення ми говоримо? Більшість із нас вдихає набагато менше диму, ніж наші предки, які користувалися буржуйками та грубками. Тому здається сумнівним, що причиною зростання алергії став зміг. Відомі випадки гострих нападів астми, спричинених сучасною побутовою хімією. Розсипані на звалищах і широко використовуються в промисловості всілякі хімікалії, такі як ізоціанати, тримелітовий ангідрид і фталевий ангідрид, потрапляють у повітря, яким ми дихаємо, і можуть бути причиною астми. Було зафіксовано, що коли починається розвантаження танкера з ізоціанатом в американському порту, поліцейські, які керують рухом поблизу, незабаром потрапляють до лікарні з нападами астми, яка потім може повторюватися знову і знову до кінця життя. І все ж є різниця між астмою, що виникла під впливом високої концентрації подразнюючої слизову оболонку речовини, і побутовою астмою, яка виникає без видимих причин. Поки немає точних даних про те, що граничні домішки хімічних речовин у повітрі можуть підвищувати ризик захворіти на астму.
Непоодинокі випадки виробничої астми у людей, які працюють на застарілих, погано обладнаних підприємствах: у звірівницьких господарствах, перукарнях, кав'ярнях, ремонтних майстернях. Описано понад 250 різновидів виробничої астми. Але набагато частіше, приблизно в половині випадків, виникає алергія на послід маленьких невидимих оком пилових кліщів, які багато копошаться в наших килимах і меблів, користуючись разом з нами благами центрального опалення.
Список алергенів, наведений Американською асоціацією легеневих захворювань (American Lung Association), гарантує нашу зустріч з одним з них, де б ми не знаходилися: пилок, пір'я, суперечки грибів, їжа, холод, емоційний стрес, надмірні навантаження, морозне повітря, пластмаси, металева стружка, дерева, вихлопні гази, сигаретний дим, фарби, аерозолі, аспірин, серцеві краплі, а в одному випадку навіть сон. Незважаючи на те, що алергенами заповнений весь світ, астма - це все ж таки переважно міська проблема. Особливо бурхливе зростання кількості хворих реєструється у нових містах, що прийшли на зміну селищам та селам. Наприклад, на південному заході Ефіопії є невелике місто Джимма (Jimma), якому трохи більше 10 років. Епідемії астми у цьому районі теж виповнилося 10 років. Причина зростання кількості алергій у містах не зовсім зрозуміла. Справді, у містах більше вихлопних газів та озону, але антисанітарні умови життя притаманні скоріше селі.
Згідно з іншою теорією астма - це результат активності клітин імунної системи, відповідальних за боротьбу з глистами. У кам'яному віці (та й у середні віки) імуно-глобулін-Е-залежна система працювала день і ніч, ведучи нескінченну боротьбу з глистами всіх пологів та різновидів. У неї не було часу дбати про екскременти кліщів та котячої вовни. Сьогодні ця система нічим не зайнята та гіперсенсибілізована на будь-які подразники. Хоча ця теорія базується на кілька сумнівних уявлень про роботу імунної системи, є
спостереження, які свідчать її користь. Немає такої гострої форми сінної лихоманки, яку не міг би вилікувати один солітер, але важко сказати, з чим пацієнт хотів би залишитися.
Ще одна теорія пов'язує зростання захворюваності на алергію в містах з тим, що люди більше часу проводять у закритих приміщеннях серед килимів та пір'яних подушок, населених багатомільйонною армією пилових кліщів. Є також теорія, згідно з якою людина стає чутливою до астми завдяки помірним вірусам (наприклад, аденовірусам, що викликають легку застуду), що вражає міське населення через його скупченість і схильність до щоденних стресів. Теорій, що пояснюють засилля вірусів, ще більше, ніж теорій виникнення астми. Тут і надмірні навантаження дітей у школі разом із переохолодженням під час змін, що вони вискакують надвір без верхнього одягу. Перманентність інфекції пояснюється тим, що зараз легко і швидко переміщаються з міста до міста і навіть із країни у країну, збагачуючи своїх співгромадян новими штамами вірусів. Відомо понад 200 різних вірусів, здатних викликати те, що ми називаємо респіраторним захворюванням. Доведено зв'язок виникнення хронічних інфекцій у дітей, а також астми з частим інфікуванням синцитіальним вірусом. Ще за однією версією виникнення астми пов'язане з її особливим впливом на імунну систему урогенітальних бактерій, що викликають неспецифічні уретрити у жінок з такою самою частотою, як і виникає астма. Ви можете вибирати будь-яку теорію, яка вам сподобалася. Особисто мені найбільш переконливою здається версія про надмірне захоплення гігієною в наші дні, втім, задля зміцнення здоров'я я все одно не житиму в стійлі. Але єдине, у чому сходяться вчені, - те, що розвиток астми обумовлено генетичної схильністю. Але як тоді бути з фактами, що свідчать про зростання числа хворих на астму? Навряд чи гени змінилися останнім часом.
То чому ж учені вважають, що астма принаймні від частини є генетичним захворюванням? Що вони мають на увазі? Приступ астми виникає внаслідок набряку дихальних шляхів під впливом гістаміну, який рясно виділяють стовбурові клітини під впливом імуноглобуліну-Е, що переходить в активний стан у присутності молекул саме тієї речовини, на яку він сенсибілізований. Ланцюжок причинно-наслідкових взаємодій прямолінійний і добре вивчений. Те, що імуноглобулін-Е може активізуватися різними речовинами у різних людей, пояснюється особливою будовою цього білка. Його просторова конфігурація може легко змінюватись під час синтезу. Як трансформатор, імуноглобулін-Е можна скрутити таким чином, щоб він ідеально входив у контакт із будь-яким чужорідним білком-алергеном. Тому в однієї людини астма може викликатися екскрементами кліщів, в іншої - кавовими зернами, але механізм розвитку реакції буде той самий - за допомогою активізації певної форми імуноглобуліну-Е.
Якщо є ланцюг біохімічних реакцій, контрольованих білками, то є і гени, що кодують ці білки. Ми пам'ятаємо, кожен білок синтезується під контролем свого гена, але у випадку з імуноглобуліном-Е це відбувається під контролем двох генів. Те, що в деяких людей алергія розвивається саме на шерсть тварин, ймовірно, пов'язане з певними змінами генів імуноглобуліну-Е внаслідок мутацій.
Це стало зрозуміло, коли з'явилися статистичні підтвердження того, що астма є сімейним захворюванням. У деяких місцях мутації, що ведуть до астми, є надзвичайно поширеними. Одне з таких місць - відокремлений острів Трістан-да-Кунья (Tristan da Cunha), населений, ймовірно, нащадками людини, яка страждала на астму. Незважаючи на приємний помірний клімат, гострі прояви астми відзначені у 20% населення острова. У 1997 році група генетиків, що фінансується біотехнологічною компанією, вирушили в далеку заморську подорож на цей острів. Були взяті аналізи крові у 270 з 300 островитян, сподіваючись знайти мутацію, що веде до астми.
Виявлення мутації зможе пролити світло на причини астми, що допоможе у пошуку нових ефективних ліків. Санітарно-гігієнічні дослідження можуть пояснити причини загального зростання захворюваності, але щоб зрозуміти, чому в одного брата розвинулася хвороба, а в іншого немає, треба знати, в якому гені сталася мутація.
Але в даному випадку, на відміну від попередніх прикладів генетичних захворювань, досить складно сказати, що є норма, а що - мутація. Що стосується алкаптонурією було зрозуміло, який ген нормальний, і який - «ненормальний». Але з астмою все набагато складніше. У кам'яному віці імунна система, що гостро реагує на пилових кліщів, не створювала проблем, оскільки пилові кліщі не були настільки поширені в тимчасовому стійбищі первісних мисливців, що нишпорять по савані. І якщо ця ж імунна система ефективно боролася з глистами, то сьогоднішній астматик був би здоровішою людиною в кам'яному віці, ніж будь-хто інший. Одним із відкриттів генетики останнього десятиліття стало те, що між нормою та мутацією не завжди є чітка відмінність.
Наприкінці 1980-х років відразу кілька груп учених розпочали пошук гена астми. До середини 1998 року знайшли не один ген, а п'ятнадцять. Вісім генів-кандидатів знаходилися на хромосомі 5, по два - на хромосомах 6 і 12, і по одному - на хромосомах 11, 13 і 14. Це не враховуючи того, що два гени, що кодують імуноглобулін-Е-центрального гравця алергічної відповіді, знаходяться на хромосомі 1. Під книгою про генетику астми могли б підписатися кожен із цих генів, причому у довільному порядку. У кожного з них були свої затяті прихильники, які лобіюють важливу роль саме свого гена у розвитку астми. Генетик з Оксфорда Вільям Куксон (William Cookson) розповідав, як його конкуренти реагували на відкриття ним зв'язку між схильністю до астми і генетичним маркером на хромосомі 11: одні вітали, інші поспішили надрукувати спростування, публікуючи результати незавершених досліджень з яв або зарозуміло висміювали «логічні диз'юнкції» та «особливі гени графства Оксфордшир». Були сказані прилюдно їдкі шпильки, а також анонімне звинувачення в підтасовуванні фактів. (Цікаво, що обман у науці вважається найстрашнішим злочином, тоді як у політиці це невинна витівка.) Навколонаукова суперечка розвивалася по спіралі - від сенсаційної публікації в Sunday,гіперболізуючою відкриття Куксона, до телевізійної програми, що зрадила обструкції публікацію, після чого була хвиля взаємних звинувачень телевізійників і журналістів. "Через чотири роки скептицизму і взаємної недовіри, - писав Куксон примирливо, - ми всі почували себе дуже втомленими" (Cookson W. 1994). Gene hunters: adventures in the genome jungle. Aurum Press, London).
Така виворітна сторона наукових відкриттів. Втім, порівнювати вчених із золотошукачами, які нишпорять у пошуках лише грошей і слави, теж було б не вірно. Через численні публікації в жовтій пресі заголовки, які повідомляють про нові гени алкоголізму або шизофренію, вже здаються поганим тоном. Закрадаються сумніви щодо ефективності самих методів сучасної генетики. Критика не безпідставна. Справді, прості та помітні заголовки в популярних виданнях не відображають всієї складності наукової проблеми. Проте вчений, який виявив зв'язок між геном та захворюванням, зобов'язаний опублікувати ці дані, не побоюючись шквалу критики та глузувань. Навіть якщо потім виявиться, що зв'язок помилковий, шкоди буде небагато - набагато менше, ніж через те, що важливий ген буде відзначений у бік через невпевненість вченого в результатах.
Куксон з колегами зрештою виявили на хромосомі сам ген і мутацію в ньому, що веде до схильності до астми. Тепер ніхто не сумнівався, що це один із генів астми. Але ця мутація пояснює лише 15% випадків захворювання. Крім того, коли інші вчені намагалися знайти підтвердження цієї залежності у своїх пацієнтів, статистична достовірність результатів була на межі помилки. Така норовлива природа всіх генів астми. У 1994 році один із суперників Куксона, Давид Марш (David Marsh), опублікував відомості про взаємозв'язок між астмою та геном інтерлейкіну-4 на хромосомі 5, виявленим при вивченні випадків захворювання у одинадцяти амських сімей.
Амські меноніт - відгалуження секти меноніт в США.
Втім, це відкриття також важко підтвердити, проводячи незалежні дослідження. У 1997 році фінські вчені переконливо показали відсутність зв'язку між цим геном та захворюванням на астму. Але в тому ж році при вивченні астми в змішаних міжрасових американських сім'ях було виявлено одинадцять ділянок хромосом, які, ймовірно, впливають на схильність до алергій. Причому десять із них були специфічними для конкретних етнічних груп. Іншими словами, гени, що впливають на схильність до астми негрів, можуть відрізнятися від генів, пов'язаних з астмою у європейців, але їх гени, у свою чергу, цілком можуть не збігатися з генами астми у латиноамериканців (Marsh D. G. 1994). Linkage analysis of IL 4 та інші хромосоми 5q31.1 маркери і загальне серце імуноглобулін-E концентрації. Science 264: 1152-1156).
Відмінності між статями виявилися не менш разючими, ніж різницю між расами. За даними Американської асоціації легеневих захворювань, вихлопні гази карбюраторних машин на бензині частіше викликають напади астми у чоловіків, тоді як для жінок більш токсичними виявилися вихлопні гази дизелів. Як правило, алергії у чоловіків виявляються в дитинстві та у юнацькому віці, але потім проходять, а у жінок – у 25-30 років, і вже не проходять. («Як правило» означає, що з цього правила є багато винятків, втім, як і з будь-яких інших.) Це спостереження пояснює той факт, що люди часто пов'язують свою спадкову схильність до алергії із хворобою матері, а не батька. Просто в батька ця схильність вже реалізувалася в дитинстві, а потім пройшла, але могла передатися дітям у спадок.
Проблема в тому, що складний механізм розвитку імунної відповіді на алергени перебуває під впливом безлічі факторів, внаслідок чого можна знайти ще багато генів астми, але вони лише частково впливатимуть на розвиток захворювання. Візьмемо, наприклад, ген ADRB 2 ,який лежить на довгому плечі хромосоми 5. Він містить пропис білка бета-2-адренергічного рецептора, під контролем якого знаходяться бронходилатація (розслаблення гладком'язових клітин повітроносних шляхів) і бронхостеноз (звуження бронхів) - дві основні ознаки астми, що призводять до утрудненого. Ліки для зняття нападу астми націлені на цей рецептор. Не дивно, що ген ADRB 2розглядався як головний претендент на назву "гена астми". Вперше послідовність нуклеотидів цього гена довжиною 1239 букв була виділена з клітин китайського хом'ячка. Потім ген був знайдений у геномі людини і зазнав ретельного обстеження. Різниця була виявлена, коли порівняли гени хворих з тяжкою формою астми з частими нічними нападами та ген хворих на інші форми астми. Відмінність полягала в єдиному нуклеотиді під номером 46. У хворих на нічну форму астми в цьому місці стояла буква А замість G. Буква G на 46-й позиції виявлялася у 8% нічних астматиків і у 52% хворих на іншу форму астми. Відмінність виявилася статистично достовірною, але не однозначною (Martinez F. D. 1997. Association між genetic polymorphism of beta-2-adrenoceptor і відповідь до albuterol в дітей з або без історії wheezing. Journal of Clinical Investigation 100: 3184-3188).
Слід зазначити, що з нічними нападами астми щодо небагато, тобто. вплив гена ADRB 2виявилося незначним. Дані інших вчених зовсім заплутали справу. Виявилося, що та сама мутація в тому ж гені впливає на звикання хворих до ліків від астми. Відомі випадки, коли ліки, наприклад формотерол, переставали діяти через кілька тижнів або місяців його застосування. Було встановлено, що звикання розвивається швидше у хворих, у яких на 46-й позиції в гені ADRB 2стоїть G замість А. Вкотре виявилося неможливо відповісти на питання, де мутація, а де – норма.
«Швидше за все», «ймовірно», «у деяких випадках» - як це не схоже на той жорсткий детермінізм, як у випадку Хантінгтонової хвороби (див. розділ 4). Безумовно, що заміна А на G, і навпаки, впливає на схильність до астми, але зовсім не пояснює, чому в одних людей розвивається астма, а в інших - ні. Вплив того чи іншого «гена астми» завжди виявлявся лише у невеликої обмеженої групи людей, тоді як в іншій групі вплив цього гена виявлявся завуальованим через безліч інших факторів. Вам слід звикати до такої невизначеності. Чим глибше ми проникатимемо в геном, тим менше в ньому буде місця для фаталізму. Генетика - гра ймовірностей, можливостей та схильностей. Не суперечить уявленням Менделя про спадковість із його простими формулами розподілу рецесивних і домінантних ознак. Просто більшість ознак перебувають під прямим чи опосередкованим впливом сотень генів, що нівелює вплив мутації щодо одного з них. Геном також складний і багатогранний, як і саме життя, тому що він і є саме життя. Сподіваюся, після цього розділу вам уже не так сумно, як після попереднього. Прямолінійний детермінізм, чи то в генетиці, чи в суспільних відносинах, діє гнітюче на тих, хто цінує свободу життя.
Матеріали представлені з навчального посібника РУДН
Анемія. Клініка, діагностика та лікування / Стуклов Н.І., Альпідовський В.К., Огурцов П.П. - М.: ТОВ «Медичне інформаційне агентство», 2013. - 264 с.
Копіювання та тиражування матеріалів без вказівки авторів заборонено та переслідується згідно із законом.
Мієлодиспластичний синдром (МДС) поєднує групу набутих захворювань кровотворної системи, при якій патологічний процес починається на рівні поліпотентної стовбурової клітини і виявляє себе порушенням проліферації та диференціювання клітин одного, двох або трьох паростків кровотворення з їхньою подальшою загибеллю в кістковому мозку (неефективний ери).
На відміну від АА, стовбурові клітини присутні в кістковому мозку хворих на МДС, хоча вони функціонально неповноцінні. Кістковий мозок при МДС частіше буває гіперклітинним, нормоклітинним і рідше – гіпоклітинним, тоді як у периферичній крові виявляється рефрактерна анемія, нерідко лейко- та/або тромбоцитопенія.
В основі функціональної патології поліпотентних стволових клітин лежать хромосомні зміни, які виявляються у більшості хворих на МДС. Вони мають клональний характер, аналогічний до цитогенетичних змін при лейкозах. Хромосомні зміни при МДС різноманітні та включають транслокацію, інверсію та делецію хромосом. До найбільш характерних відносяться: трисомія 8, моносомія 5, моносомія 7, делеція Y-хромосоми, делеція довгого плеча 7 (7q-), 11 (11q-), 13 (13q-), 20 (20q-), а також транслокації t (1; 3), t (5; 7), t (2; 11), t (6; 9), t (11; 27), інверсія 3 хромосоми. У 20% хворих спостерігаються множинні порушення. Часто зустрічається делеція довгого плеча хромосоми 5 (30% хворих). Причому встановлено, що з цим плечем 5 хромосоми втрачаються гени, що відповідають за синтез багатьох паросткових факторів, у тому числі гранулоцитарно-макрофагального, ІЛ-3, ІЛ-4, ІЛ-5, ІЛ-6 та багатьох інших біологічно активних речовин, що регулюють кровотворення .
Форма з подібною хромосомною патологією була навіть виділена серед хворих на МДС у 5
q -синдром, який частіше зустрічається у жінок, характеризується рефрактерною мегалобластною анемією та рідко трансформується у гострий лейкоз (менше 5% хворих).Причини, що спричиняють хромосомну патологію, неясні. У ряді випадків передбачається дія таких мутагенних факторів, як іонізуюча радіація, дія хімічних та лікарських факторів.
Виникла в кістковому мозку в одній поліпотентній стовбуровій клітині цитогенетична патологія, що зумовлює в подальшому розвиток МДС, здатна відтворюватися в нащадках стволової клітини, що змутувала, формуючи таким чином патологічний клон, клітини якого не здатні до нормальної проліферації і диференціації. кістковомозковою загибеллю (неефективний еритропоез). Встановлено, що 75% кісткового мозку при МДС мають
CD 95, маркер запрограмованої клітинної загибелі – апоптозу. Це зумовлює різні типи цитопеній у периферичній крові хворих на МДС.Захворюваність на МДС становить 3 - 15 випадків на 100000 населення і частота його підвищується до 30 випадків у людей старше 70 років і 70 випадків - у віці старше 80 років. Середній вік хворих – 60 – 65 років, у дітей МДС трапляється вкрай рідко.
Клініка
Клінічна картина МДС немає специфічних особливостей. Основні симптоми залежать від глибини та поєднання ураження паростків кровотворення. Основною ознакою хвороби є рефрактерний анемічний синдром, що виявляється наростаючою слабкістю, підвищеною стомлюваністю та іншими властивими анемії симптомами. У хворих на МДС з лейкопенією нерідко виникають інфекційні ускладнення (бронхіти, пневмонії та ін.). Геморагічний синдром внаслідок тромбоцитопенії спостерігається у 10 – 30% хворих, і проявляється крововиливами на шкірі та видимих слизових оболонок, кровоточивістю ясен та носовими кровотечами.
Якої-небудь характерної органної патології при МДС немає: периферичні лімфовузли, печінка та селезінка не збільшені.
Лабораторні дані
Анеміярізного ступеня вираженості спостерігається практично у всіх хворих на МДС і частіше носить макроцитарнийхарактер. Дуже рідко спостерігається гіпохромія еритроцитів. Нерідко присутні еліптоцити, стоматоцити та акантоцити, а також базофільна пунктація та тільця Жоллі в еритроцитах. У крові можуть бути ядросодержащие клітини червоного ряду. Кількість ретикулоцитів найчастіше знижена.
Часто у хворих в аналізах крові є стійка нейтропенія, причому для гранулоцитів характерна наявність псевдопельгерівської аномалії(лейкоцити з дводольчастими ядрами та дегрануляцією цитоплазми).
Тромбоцитопенія зустрічається у половини хворих на МДС. Серед тромбоцитів зустрічаються гігантські та дегранульовані форми.
У частини хворих на МДС в аналізах крові можуть зустрічатися бласні клітини.
Кістковий мозокпри МДС зазвичай гіперклітинний, але може бути нормоклітинним, а в окремих випадках – навіть гіпоклітинним. Однак, завжди є риси дисеритропоезу: мегалобластоїдність, багатоядерність еритробластів, наявність мітозів, патологічних поділів та ядерних аномалій, містків між ними, базофільна пунктація та вакуолізація цитоплазми. У частини хворих у кістковому мозку підвищено вміст сидеробластів із кільцевим розташуванням гранул заліза навколо ядра клітини.
Порушення диференціювання попередників еритроцитів при МДС проявляється підвищеним вмістом у них
HbF (рівень якого у зрілих еритроцитах нормальний) та наявністю в еритробластах пероксидази та лужної фосфатази, що є характерним для нейтрофілів.Дисгранулоцитопоез у кістковому мозку проявляється затримкою дозрівання гранулоцитів на рівні мієлоцитів, порушенням процесу грануляції цитоплазми та зниженням активності лужної фосфатази, що свідчить про їх функціональну неповноцінність, часто зустрічається гіпо-або гіперсегментація ядер нейтрофілів.
Дисмегакаріоцитопоез характеризується переважанням мікроформ та порушеною відшнурівкою тромбоцитів.
При деяких формах МДС виявляється підвищений вміст кісткового мозку бластних клітин (від 5 до 20%).
При гістологічному дослідженні кісткового мозку, отриманого методом трепанобіопсії, у ряду хворих має місце підвищене утворення ретикулінових волокон, причому різко виражений мієлофіброз спостерігається у 10 – 15% хворих на МДС. Цьому варіанту МДС, що характеризується більш вираженою гіперплазією та дисплазією клітин мегакаріоцитарного паростка, з майже 100% наявністю хромосомних аномалій, властиві більш виражена анемія, тромбоцитопенія та відносно коротка тривалість життя хворих (медіана виживання 9).
Діагностика МДС
ґрунтується на наявності рефрактерної анемії, стійкої до терапії вітаміном B 12 , фолієвою кислотою, залізом та іншими гематиками, яка нерідко поєднується з нейтро- та тромбоцитопенією та наявністю в пунктаті кісткового мозку морфологічних ознак дисгематопоезу (порушення дозрівання кровотворних клітин).Класифікація МДС:
В даний час у клінічній практиці використовуються дві класифікації: Франко-американо-британської групи (
FAB ) 1982 року та Всесвітньої організації охорони здоров'я (ВООЗ) 2008 рокам.Диференціальний діагноз
РА найчастіше доводиться диференціювати від вітамін-
B 12 - і фолієво-дефіцитних анемій, при яких також є мегалобластне кровотворення та морфологічні ознаки дисплазії клітин червоного паростка, що свідчать про неефективний еритропоез. Швидкі клінічні та гематологічні відповіді на терапію вітаміном B 12 або фолієвою кислотою вказують на причинний взаємозв'язок між анемією та дефіцитом цих вітамінів.РАКС необхідно диференціювати з набутою сидеробластною анемією, обумовленою хронічною свинцевою інтоксикацією. РЦМД, за якої є панцитопенія в периферичній крові, нагадує апластичну анемію. Наявність нормальної клітинності кісткового мозку з морфологічними ознаками дисмієлопоезу дозволяє правильно верифікувати діагноз.
Класифікація МДС (ВООЗ, 2008)
|
Нозологічна форма МДС |
Зміни у крові |
Зміни у кістковому мозку |
|
Рефрактерна анемія (РА) |
Анемія Бласти< 1% Моноцити< 1 х 10 9 / л |
- дисплазія кровотворення < 10% в одном ростке кроветворения Бласти< 5% - кільцеві сидеробласти < 15% |
|
Рефрактерна нейтропенія (РН) |
Нейтропенія Бласти< 1% Моноцити< 1 х 10 9 / л |
|
|
Рефрактерна тромбоцитопенія (РТ) |
- тромбоцитопенія Бласти< 1% Моноцити< 1 х 10 9 / л |
|
|
Рефрактерна анемія з кільцевими сидеробластами (РАКС) |
Анемія Бласти< 1% Моноцити< 1 х 10 9 / л |
- дисплазія кровотворення. Бласти< 5% - кільцеві сидеробласти > 15% |
|
Рефрактерна цитопенія з багаторостковою дисплазією (РЦМД) |
- цитопенія по 2 – 3 паростках Бласти< 1% - моноцити< 1 х 10 9 /л |
- дисплазія кровотворення < 10% в двух и более ростках кроветворения Бласти< 5% - кільцеві сидеробласти (будь-яка кількість) |
|
Рефрактерна анемія з надлишком бластів I (РАІБ-1) |
Цитопенія будь-яка Бласти< 5% - моноцити< 1 х 10 9 /л |
Бласти 5 - 9% |
|
Рефрактерна анемія з надлишком бластів II (РАІБ-2) |
Цитопенія будь-яка Бласти 5 – 19% - моноцити< 1 х 10 9 /л |
- множинна дисплазія у всіх паростках кровотворення Бласти 10 - 19% Палички Ауера ± |
|
МДС некласифікований (МДС-Н) |
Цитопенія будь-яка Бласти<1% |
- дисплазія кровотворення < 10% в одном или несколь- ких паростках кровотворення Бласти< 5% |
|
Синдром 5q- |
Анемія Бласти< 1% - тромбоцити норма або збільшено |
- нормальна або збільшена кількість мегакаріоцитів із гіпосегментованими ядрами - ізольована делеція 5q Бласти< 5% |
Гіпопластичний варіант МДС відрізнити від АА значно складніше. На користь гіпоплазії при МДС говорить наявність хромосомної патології, яка відсутня при АА, високий вміст на гемопоетичних клітинах проапоптичних білків (
CD 95) і низький рівень лужної фосфатази в гранулоцитах при МДС на відміну від нормального вмісту цього ферменту при АА.МДС з надлишком бластів відрізняється від гострого лейкозу за кількісним вмістом бластних клітин у кістковому мозку: всі випадки з бластозом більше 20% розглядаються.Лікування
Симптоматична терапія
Провідне місце у лікуванні МДС займає підтримуюча терапія, насамперед – переливання еритроцитарної маси, що супроводжується введенням десфералу або деферазироксу видалення надлишку заліза. Переливання еритроцитарної маси показано при зниженні рівня
Hb до 80 г/л і нижче, а її частота залежить від динаміки показників червоної крові. Для боротьби з геморагічний діатез використовується введення тромбоконцентрату, показання ті ж, що і при лікуванні АА. При інфекційних ускладненнях, обумовлених гранулоцитопенією, показано введення антибіотиків.Патогенетична терапія
залежить від кількості бластів до кісткового мозку. При вираженому бластозі (> 10%) необхідно регулярно проводити стернальні пункції, щоб виключити трансформацію МДС у гострий лейкоз ( acuteleukemia, AL ). При збільшенні бластів більше 20% терапія проводиться за програмами лікування AL.Алгоритм лікування МДС (Савченко В.Г., Кохно А.В., Паровичнікова О.М.)
|
Клітинність кісткового мозку |
|||
|
Гіпоклітинний кістковий мозок |
Нормо/ гіперклітинний кістковий мозок |
||
|
< 5% бластов |
5 – 20% бластів |
< 5% бластов |
5 – 20% бластів |
|
СуА |
СуА |
РЧЕПО |
Децитабін, азацитидин |
|
АТГ |
АТГ |
Спленектомія |
FLAG, 7+3 |
|
Спленектомія |
Децитабін, азацитидин |
Інтерферон-α |
МДЦ – 14 днів |
|
РЧЕПО |
МДЦ - 14 днів, 6 - МП, мельфалан |
Децитабін, азацитидин |
6 – МП |
У випадках кількості бластів у кістковому мозку стійко нижче 20% для ухвалення рішення про тактику лікування необхідно проведення трепанобіопсії, яка дозволяє встановити клітинність кісткового мозку. Після чого терапія МДС може бути спрямована на стимулювання кровотворення при гіпоплазії кісткового мозку (рекомбінантний людський еритропоетин – рч-ЕПО), імуносупресію з метою активації стовбурових клітин (АТГ, CyA ), зниження гемолізу та секвестрації клітин крові (спленектомія). При гіперклітинних варіантах або формах МДС з бластозом більше 5% лікування має включати пригнічення пухлинного росту (хіміотерапію). У Росії її найбільш підходящий алгоритм вибору терапії МДС, схема якого вказано у таблиці, сформульований фахівцями Гематологічного наукового центру: Савченко В.Г., Кохно А.В., Паровичниковой Е.Н. в 2012 році.
В останні роки для стимуляції еритропоезу у хворих на МДС, іноді успішно, використовується рчЕПО: рекормон, еритростим, епрекс, аранесп та ін., який особливо ефективний при низькій концентрації в крові нативного ЕПО (< 500 ед/мл). РчЭПО рекомендуется применять в дозе 100000 МЕ 3 раза в неделю подкожно или по 30000 – 40000 МЕ раз в неделю (при использовании пролонгированных форм эритропоэтина). Терапия считается эффективной при приросте гемоглобина более чем на 10 г/л за 4 – 8 недель или снижение зависимости от гемотрансфузий. Целевая концентрация гемоглобина 120 г/л. Через 2 месяца лечения рчЭПО сообщается о положительном эффекте у 41,6% больных с РА и у 76% больных с РАКС, причем к 6 месяцу этот эффект сохраняется соответственно у 33% и 58%. Таким образом, наиболее эффективным применение ЭПО оказалось у больных при варианте МДС-РАКС.
У білі ніж третини хворих на МДС тяжкість тромбоцитопенії може бути тимчасово знижена введенням інтерферону-α, це дозволяє уникнути алоімунізації, обумовленої введенням тромбоконцентрату.
У хворих на МДС з гіпопластичною фазою захворювання, як і при АА, ефективним виявилося проведення імуносупресивної терапії (СуА), яка не лише блокує дію Т-клітин-супресорів, а й інгібує клітинний апоптоз. Циклоспорин А призначається в дозі 5 мг/кг і викликає гематологічне поліпшення у 60 хворих цієї групи (повні ремісії розвиваються рідше, часткове поліпшення – частіше).
Для лікування форм МДС РА, РАКС, РЦМД як первинний метод лікування у літніх (старше 60 років) хворих з гіпоплазією кровотворення або при резистентності до циклоспорину в даний час широко застосовується спленектомія з біопсією печінки. Поряд із лікувальним ефектом цей підхід дозволяє виключити інші причини розвитку дисплазії кровотворення. Як правило, спленектомія дозволяє досягти тривалих перерв у гемотрансфузіях, покращити якість життя хворих.
Використання цитостатичних препаратів при РАІБ-варіанті МДС нині вважається найефективнішим лікуванням. До недавнього часу як патогенетична терапія застосовували в основному малі дози цитозару і мелфалан. Схема лікування малими дозами цитозару має такий вигляд. Підшкірно вводять по 10 мг/м 2 2 рази на день протягом 14, 21 або 28 днів залежно від кількості бластів і клітинності кісткового мозку. Мельфалан застосовують у дозах 5-10 мг/м 2 протягом 5 днів. peros . Такі курси проводять раз на місяць, як правило, півроку до 3 років, з оцінкою терапевтичного ефекту кожні 2 - 4 місяці. Ефективною терапія вважається при нормалізації або відносної нормалізації показників периферичної крові та кісткового мозку, за відсутності чи різкого зниження залежності від гемотрансфузій. Використання зазначених схем лікування призводить до розвитку часткової ремісії у 56% хворих. Однак, на виживання хворих така терапія суттєво не впливає.
При тяжкому стані хворих та неможливості проведення адекватної терапії при МДС-РАІБ-1 та -2 можна призначати 6-меркаптопурин по 60 мг/м 2 на добу. peros протягом 3 років.
В даний час робляться спроби використовувати в лікуванні МДС талідоміду та його аналога леналідоміду, позбавленого нейтротоксичної активності, але є потужним інгібітором протеаз. Застосування леналідоміду викликало зниження трансфузійної залежності у 67% хворих, причому у 58% досягалася повна незалежність від трансфузійної терапії. Варто зазначити, що цей препарат особливо ефективний при 5 q -варіант МДС, де його ефективність дорівнює 91%, тоді як при інших порушеннях каріотипу – лише 19%.
У молодих хворих до 60 років до стандартів лікування МДС-РАІБ-2 входить поліхіміотерапія. Використовують курси, що застосовуються в лікуванні гострих мієлобластних лейкоз: «7 + 3» та « FLAG ». «7 + 3»: цитарабін 100 мг/м 2 внутрішньовенно крапельно кожні 12 годин 1 – 7 дні курсу та ідарубіцин 12 мг/м 2 внутрішньовенно краплинно 1 – 3 дні курсу. « FLAG »: флударабін 25 мг/м 2 внутрішньовенно крапельно 1 – 5 дні курсу, цитарабін 2 г/м 2 внутрішньовенно краплинно 1 – 5 дні курсу + Г-КСФ (гранулоцитарний колонієстимулюючий фактор) 5 мкг/кг п/к щодня до виходу із цитопенії.
З інших активно розроблюваних препаратів у гематологічній практиці заслуговують на увагу триоксид миш'яку, бевацизумаб (авастин) та ін.
Останнім часом у клінічну практику впроваджено сучасні цитостатичні препарати інгібітори ДНК-метилтрансфераз. Механізм їх дії пов'язаний з інгібуванням процесу метилювання ДНК у клітинах пухлинного клону, що призводить до підвищення активності генів, що регулюють клітинний цикл та нормалізації процесів диференціювання клітин кісткового мозку. Дві основні речовини зареєстровані у Росії під назвою децитабін (Дакоген), азацитидин (Ведаза). За опублікованими даними найбільших міжнародних досліджень, ефективність використання цих препаратів у лікуванні МДС склала 50 – 70%. Децитабін вводять у дозі 20 мг/м 2 внутрішньовенно крапельно 1 – 5 днів на місяць. Таких курсів проводять4, далі оцінюють ефект. При позитивній оцінці продовжують терапію протягом тривалого часу до розвитку ускладнень, за відсутності ефекту використовують інші препарати. Азацитидин вводять підшкірно 75 мг/м 2 1 – 7 днів на місяць. Оцінюють ефект через півроку, далі або продовжують терапію тривалий час або змінюють препарати.
Необхідно знати, що найсерйознішим ускладненням хіміотерапії, що вимагає іноді відміни лікування, є цитопенія. Цитопенія, як правило, проявляється зниженням усіх показників крові ( Hb , лейкоцити та тромбоцити). Тяжкими станами, що загрожують життю вважається анемія менше 70 г/л, тромбоцитопенія менше 20 х 10 9 /л, лейкопенія менше 1 х 10 9 /л або нейтропенія менше 0,5 х 10 9 /л. Такі стани вимагають обов'язкового стаціонарного лікування, проведення трансфузійної та антибактеріальної терапії.
Єдиним радикальним методом лікування МДС могла б стати алогенна трансплантація кісткового мозку, проте застосування цього методу обмежується літнім віком хворих, переважна більшість яких старше 60 років.
Прогнозпри МДС залишається несприятливим і залежить від варіанта МДС. При РА трансформація у гострий лейкоз спостерігається у 15% хворих, а медіана виживання становить 50 місяців. При РАКС ці показники становлять відповідно 8% та 51 місяць; при РАІБ – 44% та 11 місяців.
 (1оцінок, у середньому: 5,00із 5)
(1оцінок, у середньому: 5,00із 5)